
 Дегенеративно-дистрофічні захворювання нервової системи з переважним ураженням периферичних нервів і м'язових волокон займають величезну частку в структурі спадкової патології людини.
Дегенеративно-дистрофічні захворювання нервової системи з переважним ураженням периферичних нервів і м'язових волокон займають величезну частку в структурі спадкової патології людини.
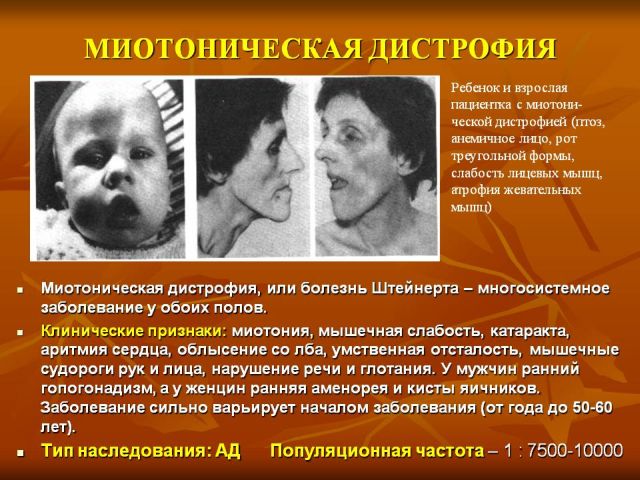
Типовим представником є міотонічна дистрофія (або дистрофічна міотонія), описана на початку минулого століття кількома авторами і отримала назву хвороби Россолимо-Штейнерта-Куршмана.
Ця недуга є найвідомішим захворюванням з розряду міотонія і найпоширенішою формою м'язової дистрофії у дорослих людей. Що являє собою ця хвороба і як з нею боротися?
Відкриття і суть захворювання
Россолимо, Штейнерта і Куршман вивчали хвороба, яка є генетичною патологією з аутосомно-домінантним типом успадкування. Це означає, що один з батьків має мутантний ген, хворі діти при цьому народжуються з імовірністю 50%. Захворювання носить характер сімейного недуги і передається наступним поколінням по вертикалі.
Сини і дочки в таких сім'ях хворіють з однаковою частотою, приблизно 3 5 осіб на 100 тисяч населення. Вік початку захворювання, а також вираженість симптомів відрізняються значною варіабельністю.
Описано ранні неонатальні й пізні форми, однак найчастіше захворювання дебютує на другому, рідше на третьому десятку життя.  Відзначено, що передача хвороби дитині від матері є більш прогностично несприятливою, ніж від батька.
Відзначено, що передача хвороби дитині від матері є більш прогностично несприятливою, ніж від батька.
В основі хвороби лежить дефект гена з 19 пари хромосом, який відповідає за синтез ферменту міотонін-протеїнкінази. Це білок в нормі присутні не тільки в скелетних м'язах, але і в клітинах міокарда та центральну нервову систему.
Ось чому для дистрофічній миотонии характерна полисистемность проявів з ураженням різних органів і систем. Неповноцінність міотонін-протеїнкінази призводить до появи м'язових спазмів разом з атрофічними змінами мускулатури голови, шийного відділу, кінцівок.
Спостерігається поєднання гіпертрофії одних м'язових волокон з атрофією інших і заміною їх на жирову або сполучну тканини.
Клінічні прояви
У зв'язку з варіюванням початку захворювання в клініці розрізняють наступні форми за віковим принципом:
- вроджена форма маніфестація хвороби починається відразу після появи дитини на світ;
- юнацький варіант дебют миотонии у віці від одного року до періоду статевого дозрівання;
- класична форма початок клінічних проявів припадає на другий і третій десяток життя;
- мінімальний варіант маніфестаціяприпадає на пізні терміни шостий десяток життя.
Характерно, що чим пізніше проявляється хвороба, тим сприятливіші протягом і краще прогноз. Найчастіше зустрічається класична форма хвороби Штейнерта, для якої типовими є наступні клінічні симптоми:
- Міотонія проявляється спазмами жувальних м'язів і згиначів кистей рук, характерні атрофічні зміни в різних
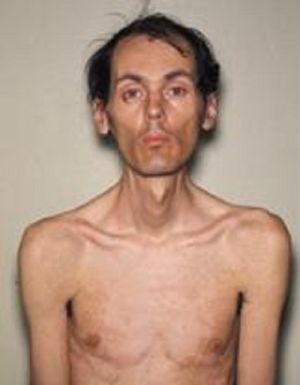 групах м'язів. Поступово відбувається згасання міотоніческім проявів і прогресування м'язової дистрофії, зовні це виражається в сумної масці особи і відсутності міміки. Небезпечним є парез м'язів гортані з порушенням ковтання, а також слабкість дихальної мускулатури, в результаті можливі напади зупинки дихання уві сні, розвиток пневмонії.
групах м'язів. Поступово відбувається згасання міотоніческім проявів і прогресування м'язової дистрофії, зовні це виражається в сумної масці особи і відсутності міміки. Небезпечним є парез м'язів гортані з порушенням ковтання, а також слабкість дихальної мускулатури, в результаті можливі напади зупинки дихання уві сні, розвиток пневмонії. - Серцево-судинні порушення порушення ритму серця, гіпертрофічні зміни лівого шлуночка, які виявляються на ЕКГ, застійна серцева недостатність.
- Ендокринні розлади (в основному зачіпаються статеві функції) зменшення розмірів статевих органів, зниження сексуального потягу, у жінок розлади менструального циклу, ожиріння.
- загальні зміни дистрофічного характеру сухість і пігментація шкірних покривів, випадання частково або повністю волосся і зубів, рання катаракта.
- Порушення з боку ЦНС втома, розлади сну, апатія, втрата інтелекту.
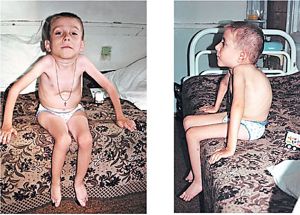
Окремо варто відзначити характерні клінічні прояви вродженої форми дистрофічній :
- зменшення активних рухів плода в утробі матері, що виявляється під час УЗД;
- в період новонародженості млявість, поширена , особливо в жувальних, мімічних, м'язах очних яблук;
- збереження і навіть підвищення сухожильних рефлексів;
- проблеми вигодовування, розлади дихання за типом респіраторного дистрес-синдрому;
- затримка фізичного і нервово-психічного розвитку, ознаки олігофренії;
- стрімке прогресування захворювання, високий ризик раптової смерті.
Діагностичні критерії
Підозра на хворобу Россолимо-Штейнерта Куршмана може виникнути у лікаря при наявності у пацієнта поєднання міотоніческім і дистрофічних змін в м'язах на тлі втрати інтелекту та наявності серцево-судинної і ендокринної патології.
Полисистемность практично завжди свідчить про генетичну природу захворювання. Такі хворі підлягають аналізу ДНК і проведення генеалогічного аналізу для підтвердження аутосомно-домінантного успадкування патології. Як інформативних методів дослідження використовуються електрокардіографія, , аналізи на гормони.
У зв'язку з багатогранністю клінічних проявів до процесу постановки діагнозу зазвичай залучаються фахівці з різних галузей медицини генетики, кардіології, ендокринології, гінекології , андрології, неврології.
Диференціальний діагноз проводиться між дистрофічній міотонію і іншими видами схожих захворювань. На відміну від інших для хвороби Россолимо характерна м'язова атрофія. Нерідко для підтвердження діагнозу доводиться вдаватися до біопсії, щоб визначити рівень м'язового білка, який в тканинах при даній патології підвищений.
Проводиться також антенатальная діагностика методом дослідження навколоплідних вод.
Медична допомога
Генетичне захворювання неможливо вилікувати повністю, тому метою лікування при хворобі Россолимо-Штейнерта-Куршмана є купірування симптомів, поліпшення загального стану і соціальна адаптація пацієнтів.
Принципи лікування полягають в наступному:
- дієта з низьким вмістом солей калію (яблука, спаржа, капуста, огірки, виноград, зелень, кукурудза, ягоди , редис, мандарини,
 грейпфрут, цибулю, моркву, баклажани, горох);
грейпфрут, цибулю, моркву, баклажани, горох); - виключення переохолоджень , щоб уникнути виникнення спазмів;
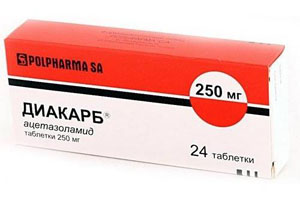
- застосування препаратів хініну для стабілізації клітинних мембран, таких ліків, як Дифенін, Новокаинамид, Диакарб для зняття м'язових спазмів і зменшення скутості, судом, зниження внутрішньочерепного тиску;
- використання анаболічних стероїдів (Метанандростенолон, Ретаболил, нерабол), вітамінів групи В, АТФ для стимулювання м'язової маси;
- , масаж , електроміостімуляція, ортопедичні пристрої .
Перераховані заходи дають хороший позитивний ефект як при класичній, так і при вродженої формі хвороби. Повністю позбавити хворого від дистрофічній миотонии вони не можуть, але продовжити йому життя і поліпшити її якість здатні.
Прогноз гірше у вродженої форми летальність висока, діти можуть не дожити до 3 років. Юнацький варіант миотонии протікає досить 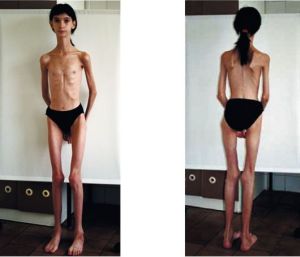 важко і може привести вже в молоді роки до обмеження працездатності та ранньої інвалідності.
важко і може привести вже в молоді роки до обмеження працездатності та ранньої інвалідності.
У разі класичної форми захворювання може протікати довго при проведенні своєчасних лікувально-корекційних заходів. Найбільш сприятливий прогноз у пізно проявилися форм хвороби.
Профілактичні заходи зводяться до того, що жінці з родини з неблагополучним анамнезом на стадії планування вагітності необхідно пройти обстеження на наявність аномальних генів, відповідальних за розвиток м'язової дистрофії. Це доцільно зробити також у разі наявності у родичів батька дитини даної патології.
Можливості до народження діток повинні вирішуватися індивідуально в кожному конкретному випадку лікарями генетиками після консиліуму.