
 Синдром Айкарді є рідкісним цереброретінальним генетичним розладом. При цьому захворюванні мозок, частково або повністю, позбавлений однієї з основних складових — мозолистого тіла. Це є наслідком дефекту Х-хромосоми. У сітківці відбуваються зміни, що ведуть до інфантильним спазмів.
Синдром Айкарді є рідкісним цереброретінальним генетичним розладом. При цьому захворюванні мозок, частково або повністю, позбавлений однієї з основних складових — мозолистого тіла. Це є наслідком дефекту Х-хромосоми. У сітківці відбуваються зміни, що ведуть до інфантильним спазмів.
Мозолисте тіло — центральний відділ нервової системи, представлений нервовими закінченнями, що з'єднують півкулі головного мозку.
Сидром Айкарді не є окремою формою , а є самостійним захворюванням, яке в 1965 році було описано невропатологом з Франції Жаном Екарда.
Синдром Айкарді-Гутьєреса є загрозою для хлопчиків з хромосомним набором ХХY або з синдромом Клайнфельтера.
За даними статистики, на сьогоднішній момент спостерігалося близько 500 випадків цього захворювання. Найбільшого поширення воно отримало в Японії.
Етіологія і патогенез синдрому
До сих пір точно не встановлена причина даного захворювання. Відповідно, неможливо визначити фактори ризику.
Фахівці відкидають спадковість. Якщо в сім'ї є дитина з даними синдромом, то шанс народження другого з такою ж хворобою становить менше 1%. 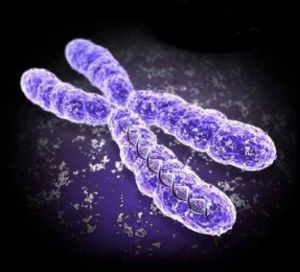
Було висунуто наступна теорія. Клітини ембріонів містять по одній активної Х-хромосомі. Отже, їх інактивація при нормальному розвитку відбувається випадково.
При синдромі Айкарді, випадкової інактивації не спостерігається і більшість клітин формується за участю тільки цієї хромосоми.
Який ген відповідає за ці зміни до теперішнього часу невідомо . Імовірно, це мутований de novo на інактивованої Х-хромосомі.
Як проявляє себе синдром?
Діти 2 — 5 місяців, у яких виявляється синдром Айкарді, зовні не відрізняються від здорових малюків. Пізніше починають проявлятися судоми або інфантильні спазми. Вони представляють вид .
Дитина знижує активність діяльності і впадає в ступор. Погляд прикутий до однієї точки. Руки починають підніматися вгору і згинатися.
Тіло вигинається і ніжки починають різко випростатися. На це йдуть секунди і продовження можна очікувати будь-якої миті. Під час нападів, малюк стає дратівливим і постійно плаче. Пізніше, такі судоми часто ведуть до епілепсії.
Паралельно присутні і інші симптоми синдрому Айкарді:
- уражається сітківка, в очах з'являються жовтуваті плями;
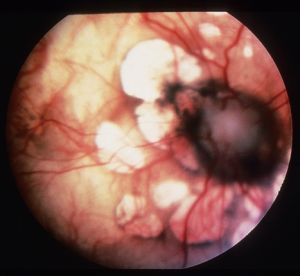
На фото очне яблуко при синдромі Айкарді
- очі дитини ненормально-малого розміру;
- вроджена колобома — виїмка, щілину або проріз в райдужній оболонці ока;
- затримка розвитку;
- проблеми при годуванні дитини;
- діарея;
- потрапляння їжі і шлункового соку в стравохід — гастроезофагеальнийрефлюкс (у дорослих-печія). Часто проходить самостійно і не є серйозним захворюванням. Однак слід знати, що це одна з ознак захворювання;
- м'язова апатичність.
За спостереженнями за хворими можна виділити і додаткові ознаки:
- латеральное розташування брів, їх рідкість;
- виступаючі різці та кирпатий ніс;
- зменшений кут носової перегородки.
Іноді спостерігаються нарости і ущільнення на шкірі: невуси, дивертикули шкіри і пухлини, причиною яких є патологія кровоносних судин — гемангіоми. Рідко зустрічається аномалія рук.
Зустрічаються хворі з плагіоцефаліі (сплюснута область на голові), з асиметрією особи, а також з ущелинами неба або верхньої губи. Розміри голови і рук помітно менше, ніж у здорової людини. Ніс плоский, а вуха надмірно великі. Ніс і губи розділяє занадто малий простір.
У більшої кількості хворих відбувалося розвиток важких , які не проходили до кінця життя. Відомо наявність напів-хребців і відсутності ребер, що приводить до помітного викривлення хребта.
Збільшено ризик розвитку пухлин:
- папілом судинного сплетення;
- ліпом;
- ангісарком;
- гепатобластому ;
- гемангіом;
- ангіосарком;
- гепатобластому;
- интерстициального поліпозу;
- ембріональної кальціоми;
- поліпозу кишечника.
 Аномалії в очах можуть призвести до пігментному Реніта, мікрофтальм. Можлива катаракта і атрофія зорового нерва. Це може привести до порушення зору і навіть повної сліпоти.
Аномалії в очах можуть призвести до пігментному Реніта, мікрофтальм. Можлива катаракта і атрофія зорового нерва. Це може привести до порушення зору і навіть повної сліпоти.
Нерідко порушується ендокринна система — пізнє настання статевої зрілості або явна затримка.
Фізичний розвиток починає сповільнюватися. У віці 7-10 років хворий виглядає як п'ятирічний. Вага збільшується також із затримкою.
Значна кількість хворих мають явно виражену затримку в розумовому розвитку, однак, іноді вони просто погано здатні до навчання.
Здатність до членороздільноюмови часто розвинена дуже слабо. Самостійне пересування спостерігається рідко. Деякі повністю залежні від допомоги інших людей.
Постановка діагнозу
На сьогоднішній момент немає спеціальної технології діагностики, за допомогою якої можна точно поставити діагноз. Як правило, застосовуються такі методи:
- Неврологічний огляд , в процесі якого лікар оцінює стан нервової системи дитини і визначає її розлади.
- офтальмоскопія включає в себе вивчення очного дна, його судин і сітківки.
- (ЕЕГ) . З її допомогою досліджують роботу головного мозку, відбувається реєстрація електричних імпульсів, які виходять з його окремих частин. Є основним методом для діагностики епілепсії та інших захворювань. При синдромі Айкарді допомагає визначити тяжкість і тип нападів.
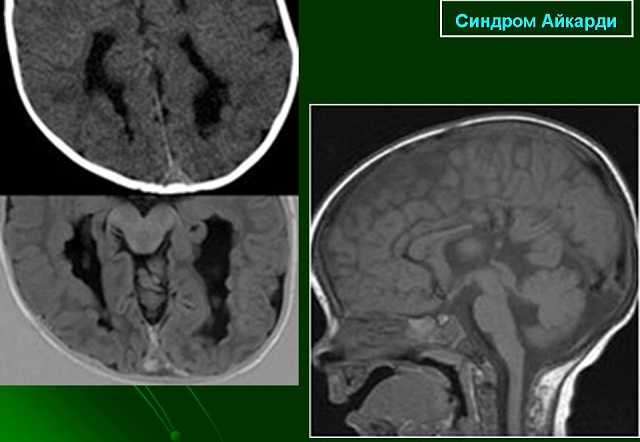
- (МРТ) . Основу цього діагностичного дослідження становить постійне магнітне поле, швидко мінливі локальні магнітні поля і радіочастотна енергія. Спеціально призначена для цього апаратура створює чітке зображення внутрішнього органу. За допомогою виявляється патологія мозолистого тіла, асиметрія півкуль, внутрішньомозкова кіста і інші аномалії.
- (КТ) . З її допомогою фахівці виявляють ділянки мозку, які зазнали пошкоджень в результаті хвороби.
- Рентген скелета .
- Генні дослідження .
Інші діагностичні методи можуть проводитися в залежності від симптомів і загального стану хворого.
Симптоматичне лікування єдиний варіант
На сьогоднішній момент комплексного лікування синдрому Айкарді немає. Застосовується терапія кожного симптому.
Прийнята тактика — купірувати інфантильні спазми, які є резидентними до антиепілептичних препаратів. Використовуються максимально високі дози різноманітних медикаментів.
Після виявлення захворювання, на перших порах використовується Вігабатрін, він же Сабріл. Його застосовують від 50 до 100 мг на кілограм ваги в добу. Паралельно з ним вводяться вальпроати. Їх добова норма — 50-100 мг на кг.
Якщо напади частішають, то до АЕП додаються бензодіазепіни, від 0,25 до 2 мг на добу. Нерідко застосовується Фенобарбітал або Суксилеп.
В якості альтернативи виступають кортикостероїдні гормони. Це:
- АКТГГ;
- Синактен-депо;
- Дексаметаз;
- Преднізолон;
- Октагам .
Спочатку преднізалона призначається з розрахунку 1-2,5 мг на кг, потім ця доза знижується до делікатної. Як правило, гормони застосовуються паралельно базовим АЕП.
На жаль, ці методи малоефективні і рідко досягають повного придушення синдрому.
У вигляді паліативної хірургічного лікування може бути стимульований блукаючий нерв. Дефекти кісток, які сприяють сколіозу, усуваються фізіотерапією, лікувальною фізкультурою або виправляються за допомогою хірургічної корекції.
Розроблено спеціальні програми, які протидіють розумової відсталості. Для тривалого лікування патологічних станів необхідна допомога дитячого невропатолога.
Прогноз і летальність
Прогноз при синдромі Айкарді досить несприятливий і залежить від тяжкості спазмів і супроводжуючих їх захворювань:
- смертність в дитячому віці — 25%;
- виживають і можуть самостійно ходити — 25%;
- можуть самостійно себе обслуговувати — 50%.
Хворі живуть приблизно від 9 до 19 років. Однак відомі випадки, коли хворі жінки прожили до 32 і 49 років (помірна форма синдрому).
Незважаючи на рідкість захворювання, йому присвячений окремий сайт (). На ньому можна поспілкуватися з іншими людьми, страждаючими від цього захворювання, і їх сім'ями. Задати питання, поділитися досвідом і інше. Тут можна отримати інформацію про благодійні заходи, організатором яких є Фонд Екарда.
На жаль, у зв'язку з мало вивченою даного захворювання, профілактичних заходів проти синдрому Айкарді поки не існує.