
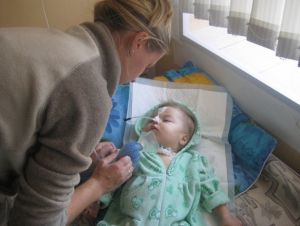
 Хвороба Александера є дуже рідкісну форму лейкодистрофии.
Хвороба Александера є дуже рідкісну форму лейкодистрофии.
Це одне з нейродегенеративних захворювань нервової системи. При розвитку цієї недуги відбувається руйнування білої речовини — мієліну .
Цей компонент необхідний організму для утворення мієлінової оболонки нервових волокон, а також для передачі нервових імпульсів.
При захворюванні спостерігаються комбіновані порушення в роботі нейронів і імунних клітин.
Зміст
Форми захворювання
Подібним захворюванням найчастіше страждають діти. Хвороба Александера зазвичай призводить до летального результату протягом 10 років після того, як з'явилися перші симптоми .
На даний момент розрізняють три форми недуги:
- дорослу ;
- ювеніальную;
- інфантильну.
Крім цього фахівці визнають неонатальную форму. В даному випадку хвороба виникає у дитини, що знаходиться в утробі матері в період виношування. Кожна з форм має виключно генетичну основу.
Потенційна група ризику
До основної категорії людей, які страждають від цього захворювання, відносяться діти з генетичної схильність . Кожен зафіксований випадок закінчився летальним результатом.
На даний момент відомо не так вже й багато випадків хвороби Розенталя. Саме з цієї причини захворювання ще не до кінця вивчений. Як показують дослідження, недуга може з'явитися у людини в будь-якому географічному розташуванні, будь-якої статі і раси.
Причини розвитку хвороби
Вчені довели, що лейкодистрофия Александера розвивається в результаті змін, що відбуваються в 17 хромосомі і що носять мутаційний характер. 
На довгому плечі саме цієї хромосоми розташований ген. При порушеннях в ньому руйнується кислий гліальний білок.
Однак існує невеликий відсоток людей, які страждають цим захворюванням, у яких даний ген в повному порядку. На жаль інші причини розвитку хвороби Александера досі невідомі.
Крім цього дослідження показали, що подібні зміни в 17 хромосомі не передаються у спадок. Вони виникають спонтанно при формуванні генетичних даних дитини.
Це доводить, що жоден з батьків не є носієм подібного мутаційного гена.
Імовірність повторного народження дитини з подібними відхиленнями дорівнює нулю. Вчені припускають, що подібні зміни можуть відбуватися в процесі утворення сперматозоїдів саме на батьківській хромосомі.
При цьому подібні мутації абсолютно не залежать від віку.
Особливу роль в процесах розвитку нервових клітин відіграє кислий білок — GFAP. Однак змінений ген провокує накопичення в клітинах нейроглії, які беруть участь в проведенні в вигляді імпульсів нервові сигнали, більшого кількість білка з порушеною структурою.
Крім цього клітинами гліальних тканини забезпечуються всі нейрони необхідними корисними компонентами. Іншими словами, підтримується їх життєдіяльність.
При наявності хвороби Александера у пацієнта в кислому білку відбувається стрімке накопичення вузликів — Волокон Розенталя. Згодом вони заважають проходженню по астроцитам імпульсів, що призводить до порушень в роботі всієї ЦНС.
Діагностування захворювання
Первинним показниками для постановки діагнозу служить збільшення розмірів голови пацієнта.
Фахівці в обов'язковому порядку повинні провести ряд обстежень хворого.
Для цього зазвичай застосовують КТ, . Крім цього необхідно провести ряд аналізів, які допоможуть виявити можливі захворювання з лейкодістрофіческого ряду.
Остаточний же діагноз лікар може поставити тільки після вивчення отриманих результатів та аналізу крові, який зобов'язаний виявити у пацієнта зміни ДНК мутаційного характеру, а також ідентифікувати кислий гліальний білок.
Звичайно, останній пункт навіть при відсутності або ж наявності зміненого білка не гарантує точність даних і не підтверджує, що це хвороба Александера.
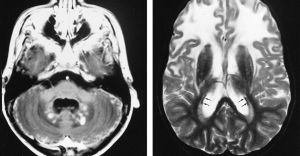 Найчастіше при негативних показниках тесту після розтину у пацієнта виявляється повна руйнація мієліну або ж волокна Розенталя.
Найчастіше при негативних показниках тесту після розтину у пацієнта виявляється повна руйнація мієліну або ж волокна Розенталя.
Варто відзначити, що такі вузлики здатні грунтовно пошкодити подкорковую частина головного мозку, а також спинний мозок. Волокна оточують капіляри, зачіпаючи при цьому м'яку оболонку мозку.
Складність виявлення симптомів
Так як захворювання зустрічається досить рідко, детально вивчити, а головне, виявити його симптоми дуже складно. До того ж для кожної форми недуги характерні різні ознаки, які залежать від умов протікання хвороби.
неонатальна форма
Якщо зміна хромосоми відбулося в неонатальної формі ще в утробі матері , то смерть немовляти настає в період першого року життя.
При цьому діти народжуються відразу ж з набряком мозку. Звичайно ж, значно підвищується внутрішньочерепний тиск і спостерігається зупинка в нервовому розвитку дитини.
Перші роки життя
Інфантильна форма захворювання зазвичай виникає в перші кілька років життя дитини.
Ознаками хвороби Александера в даному випадку служить затримка у фізичному і психологічному розвитку, що в результаті призводить до повної асоциализации.
Ювенальний період
На фото дитина у якого хвороба Александера
Приблизно 20% хворих страждає ювенальної формою недуги, яка виникає зазвичай до 10 років.
В даному випадку у пацієнта може спостерігатися задишка, порушення ковтальних рефлексів і правильно говорити, інтелектуальна деградація, утрачивание пам'яті.
При цьому сильне пошкодження доводиться на стовбур головного мозку. Поступово відбувається руйнування мієліну.
Ознаки у дорослих
Що стосується дорослої форми, то вона сильно варіюється в симптоматиці. До перерахованих вище ознаками хвороби Александера варто додати мимовільне рух очних яблук.
Це вказує на затримку психічного розвитку хворого. При цьому пацієнт досить часто мучиться, в ранковий час особливо, від блювотних поривів.
Якщо у людини діагностовано мозочкова атаксія, то він втрачає можливість нормально пересуватися і тримати рівновагу. і його профілактика.
Лікування симптомів
На жаль, сучасна медицина не має у своєму арсеналі ефективного препарату проти хвороби Александера. Цілком ймовірно, що майбутнє саме в цьому напрямку залежить від розвитку генної інженерії.
Після постановки діагнозу хворому призначають симптоматичне лікування, яке дозволяє значно полегшити стан пацієнта і продовжити його життя:
- Для зменшення мимовільних рухів використовують нейролептики, наприклад, Азалептин, і інші.
- При епілептичних припадках можуть призначити протисудомні засоби, наприклад, Сибазон, Вальпроати та інше.
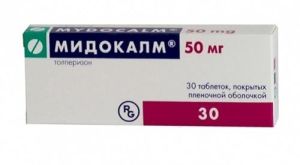
- При спастичності м'язів використовують міорелаксанти, наприклад, Мідокалм, Сирдалуд, Баклофен.
Для пересування можуть бути використані спеціальні пристосування.
у пік захворювання пацієнт може переміщатися тільки в інвалідному візку. У термінальних стадіях хворий потребує стороннього догляду.
Невтішний висновок
Хвороба Розенталя є рідкісним, генетичним захворюванням. Розгорнута клінічна картина в даному випадку представлена координаторні, руховими порушеннями, а також проблемами, пов'язаними з прийомом їжі і промовою.
Велика частина людей з подібним захворюванням живе близько 10 років. Найточніший метод діагностики недуги — генетичний. Що стосується методів лікування, то вони знаходяться в стадії розробки.
На даний момент хворим допомагають засоби, спрямовані на зняття симптомів.