
 СМА або спінальна м'язова атрофія — це група з декількох видів спадкових патологій, в результаті яких відбувається часткова або повна втрата рухових нейронів спинного мозку в передній частині.
СМА або спінальна м'язова атрофія — це група з декількох видів спадкових патологій, в результаті яких відбувається часткова або повна втрата рухових нейронів спинного мозку в передній частині.
Дана проблема відбувається з- за мутації ДНК гена SMN1, який продукує білок, через мутації кількість білка у хворих людей нижче норми, а це гарантована втрата моторних нейронів.
Для даного захворювання характерне порушення роботи поперечно-смугастої м'язової ніг, голови і шиї. Хворим погано вдається довільно пересуватися:
- повзати;
- ходити;
- утримувати голову;
- ковтати.
Руки при цьому працюють відмінно, присутній чутливість і хороші психічний розвиток.
Зміст
Історичні факти
У 1891 році СМА у дітей вперше описав вчений Вердніга. Він чітко представив опис патоморфологічних змін м'язів, різних  груп, периферичних нервів і спинного мозку, при цьому він зазначив атрофію клітин передніх рогів і корінців спинного мозку.
груп, периферичних нервів і спинного мозку, при цьому він зазначив атрофію клітин передніх рогів і корінців спинного мозку.
У 1892 році Хоффман відзначив нозологическую самостійність проблеми .
У 1893 році Вердніга і Хоффман надали докази дегенерації клітин рогів спинного мозку.
У 1956 році була представлена загального обговорення пізніша форма СМА, що володіє більш позитивними прогнозами.
Про СМА є і пізнавально
Цікаві факти про спінальної м'язової атрофії:
- незважаючи на рідкість даної проблеми, це найпоширеніше порушення генетичної форми;
- в дитячому віці захворювання передається по аутосомнорецессівний лінії;
- даний ген картирован на 5-й хромосомі і ідентифікований в 1995 році;
- серед діток середній рівень народжуваності з даною проблемою 1 на 6000 — 10000 чоловік;
- дітки 1 форми м'язової атрофії не доживають до 2-х років в 50% випадків;
- СМА може проявитися не тількипри народженні, але і в середньому або літньому віці;
- за середніми показниками кожен 50 людина володіє рецесивним геном;
- дитина, у якого є два даних гена, і з боку матері, і з боку батька на 25% схильний до поразки, для нормального самопочуття досить мати 1 нормальний ген.
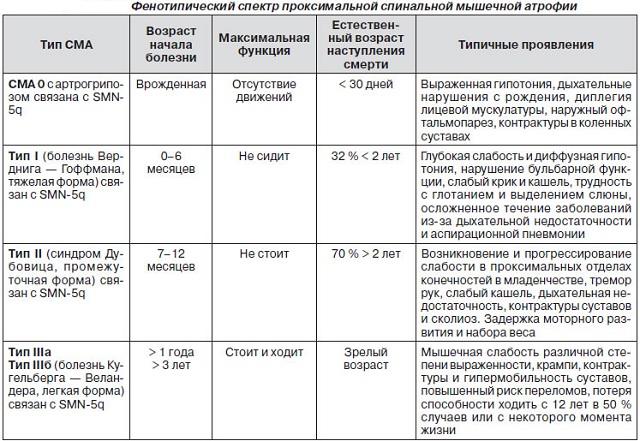
Класифікація і симптоми за типовими відмінностей
Розрізняють декілька типів СМА.
Аміотрофія Верднига-Гоффмана
I тип — дитячий, , до півроку. Дуже важкий вид, такі дітки самостійно не сидять, голова не тримається, труднощі з смоктанням, ковтанням і диханням, проявляється фасцикуляція мови, .
 Хвороба помітна ще з перших годин народження, відсутністю глибоких рефлексів і вираженою м'язовою гіпотонією. Яскраво виражені кістково-суглобові деформації. Навіть у тієї невеликої кількості діток, які зможуть сісти і тримати голову ці навички скоро регресують.
Хвороба помітна ще з перших годин народження, відсутністю глибоких рефлексів і вираженою м'язовою гіпотонією. Яскраво виражені кістково-суглобові деформації. Навіть у тієї невеликої кількості діток, які зможуть сісти і тримати голову ці навички скоро регресують.
Часто у таких діток існує порок розвитку тазостегнового суглоба, клишоногість, наявність «курячої» грудної клітки.
Прогноз : захворювання швидко прогресує, злоякісна форма. Смерть відбувається до 9-річного віку, здебільшого через проблеми з серцево-судинною системою і дихальної недостатності.
Хвороба Дубовиця
II тип — проміжний, хвороба Дубовиця, до 18 місяців. Такі дітки вже пройшли сприятливо перші етапи розвитку, однак вони не зможуть навчитися ходити, розвиток хвороби залежить від ступеня патології респіраторних м'язів.
Зазвичай симптоми спінальної м'язової атрофії цієї форми проявляються в підгострому перебігу після перенесеного стресу для фізичного здоров'я, починається з нижніх кінцівок і поступово переходить на верхні ділянки.
З'являється фасцикуляція мови, невеликий , .
Прогноз : більш м'який перебіг злоякісного характеру, летальний результат відбувається у віці 14 — 15 років.
хвороба Кюгельберга-Веландер
III тип — юнацький, хвороба Кюгельберга-Веландер, від 18 до 30 місяців. Цей тип менш схильний до смертельного результату, дитина

На фото програміст Валерій Спиридонов з Росії діагноз спінальна м'язова атрофія
самостійно стоїть, однак цей процес для нього дуже хворобливий, надалі він буде інвалідом, без коляски або тростини не пересувається.
Починається хвороба непомітно, малюк починає ходити ходою «заводний ляльки», спотикається і падає, рухи стають невпевненими. Атрофія м'язів спочатку погано помітна через присутність розвиненого підшкірного жиру.
Однак з'являються все ті ж симптоми , фасцікуляціі мови, . Вже на ранніх стадіях захворювання зникають добре розвинені рефлекси.
Відбувається деформація суглобів і кісток, особливо це помітно в області грудної клітини.
Прогноз : дітки втрачають здатність ходити до 10 — 12 років, а живуть до 20 — 30 років.
Захворювання в дорослому віці
IV тип — дорослий, після 35 років. Через атрофії сухожильних рефлексів, зниження проксимальної мускулатури, людина втрачає можливість самостійно пересуватися.
Прогноз : протягом не має впливу на тривалість життя.
Інші види та типи СМА
Крім спінальної м'язової атрофії, що передається на генетичному рівні, існує ще певна кількість споріднених хвороб:
- 1 типу Х-зчеплена аміатрофія — початок в 40 — 60 років;
- 2 — вроджена, дуже агресивна форма, призводить до смерті до 3 місяців;
- 3 — уражаються всі кінцівки, майже у всіх випадках страждають хлопчики;
- дистальная спінальна аміатрофія 1 типу — з самого народження, іноді внутрішньоутробно, вражені верхні кінцівки;
- 2 і 4 типу — це захворювання було описанотільки в одній сім'ї;
- 3 — повільно розвивається захворювання;
- 5 типовий характер — повільно розвивається в молодому віці;
- VA і VB-типу — ураження верхніх кінцівок;
- ураження гомілок — з'являється в юнацтві, повільно розвивається зі слабкості в колінах;
- поразку голосових зв'язок — параліч зв'язок у дорослих;
- аутосомно-домінантна спінальна аміатрофія — з'являється в дорослому віці, а її ювенільний форма в юнацтві;
- вроджений характер — захворювання з перших моментів життя;
- для лопатки малогомілкової вид — дужерідкісне захворювання нижніх кінцівок з викривленням стопи;
- ювенильно сегментальний вид ураження спинного мозку — дуже рідкісний випадок, проявляється в юності з прогресом 2 — 4 роки, після уражаються руки;
- хвороба Фенкеля — розвивається в основному в районі 37 років, але присутній випадки появи в 12-річному віці;
- хвороба Джокеля — пізніше і повільний початок;
- спінальна аміатрофія з міоклонічні епілепсію — уражаються кінцівки, по здебільшого в слідстві миоклонических нападів;
- спінальна форма з вродженими переломами кісток — важкаформа м'язового виснаження;
- понтоцеребеллярная гіпоплазія — існує її 8 видів;
- асиметрична сегментальная форма — хвороба 15 — 25-річних людей.

Диференціальний аналіз
Прогнозування можливо завдяки отриманим даними в результаті:
- генетичного аналізу;
- особливостей клініки (тип хвороби, місце локалізації);
- супутні симптоми, наприклад, посмикування мови;
- результатах ЕКГ і даних біопсії скелетних м'язів.
Диференціювати дитячий і ранній вид СМА можна на підставі проблем з вродженою м'язовою гіпотонією — синдрому «млявого дитини», аміатоніі, вродженого характеру доброякісної дистрофії м'язів, спадковості і хромосомного аналізу.
Юнацький вид відрізняється від спінальної аміатрофіі Кугельберга-Вееландер і різних видів м'язової дистрофії.
Завдання і методи лікування
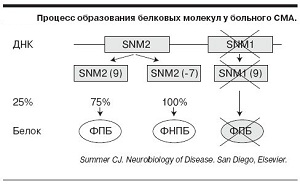 Так як дане захворювання відбувається здебільшого з -за нестачі SMN-білка, основне завдання лікування полягає в підвищенні його рівня. На сьогодні в цьому напрямку ведуться активні дослідження в інститутах США, Італії, Німеччини.
Так як дане захворювання відбувається здебільшого з -за нестачі SMN-білка, основне завдання лікування полягає в підвищенні його рівня. На сьогодні в цьому напрямку ведуться активні дослідження в інститутах США, Італії, Німеччини.
Завдяки групі добровольців проводиться клінічне дослідження терапії СМА вальпроєвої кислотою, бутіран натрієм та іншими медикаментами. Дані про застосування стовбурових клітин поки не оприлюднені.
На стан хворих позитивно діють:
- масаж;

- нейром'язова стимуляція;
- фізіопроцедури, будь-які фізичні навантаження несуть тільки позитивний ефект.
- хворі СМА потребують дієті;
- курсової прийом препаратів, до складу яких входять вітамінні комплекси, знеболюючі ліки, що покращують метаболізм нервової тканини і м'язів, стимулюють їх.
На сьогодні стан хворого можна підтримати і трохи полегшити, але спінальна м'язова атрофія залишається невиліковною.
Профілактика
Профілактичні заходи полягають в пасивних діях — консультації майбутніх батьків, у яких є ген СМА про можливий ризик, пренатальна ДНК-діагностика до 14 тижнів вагітності, біопсія ворсин хоріона для постанови остаточного діагнозу і прийняття рішення батьками про подальші дії.