
 У 2001 році даний синдром був внесений до переліку хвороб, для яких властиві епілептична активність і епілептиформні порушення в .
У 2001 році даний синдром був внесений до переліку хвороб, для яких властиві епілептична активність і епілептиформні порушення в .
Ці порушення викликають розвиваються погіршення стану мозку . Також була прийнята гіпотеза Отахара про те, що дана хвороба в 80% ситуацій переростає в іншу — . А ще через час бували ситуації з трансформацією хвороби в .
Синдром Отахара — це початкова стадія епілептичної енцефалопатії, яка виявляється у дітей в перші 3 місяці після народження. Для хвороби властиві гострі напади, які з'являються протягом 10-денного життя дитини, в рідкісних випадках після появи на світло.
Порушення в метаболізмі немовляти говорять про сімейні захворюваннях. Виявляється загострено на тлі ідеального здоров'я.
Причини і етіологія хвороби
Найпопулярнішою передумовою вважаються вади формування головного мозку — унилатеральной мегаленцефалія, і інші. Іноді в якості джерела можуть виступати картіровочние порушення, збої пов'язані з обміном речовин.
У персональних дослідженнях Отахара розглянув 10 ситуацій. В результаті у 2 була присутня кіста в півкулях мозку поренцефалія,  також у 2 і підгостра змішана — дистрофічні зміни тканин мозку, що і викликає порушення його функцій . У інших 6 дітей походження хвороби не виявлено.
також у 2 і підгостра змішана — дистрофічні зміни тканин мозку, що і викликає порушення його функцій . У інших 6 дітей походження хвороби не виявлено.
У ще одному обстеженні з 11 немовлят, у 1-го була асфіксія при народженні, у 1-го з вродженою патологією, важливу цінність у розвитку і поширенні якого грає генетика ( ).
у 1-го некетоновой гіпергліцінемію, у інших підстави хвороби не виявлено. Тільки 1 був з подібними симптомами епілепсії як і у його родичів.
Schlumberger в своєму експерименті в 8 з 8 ситуацій поставив діагнози, пов'язані з вадами головного мозку. 6 з яких — унилатеральной мегаленцефалія і один синдром Айкарді. Унилатеральной мегаленцефалія як причина зустрічалася ще в дослідженнях Martin, Ohtsuka.
У 1995 році в статті, присвяченій дитячої епілепсії, було написано, що джерелами синдрому Отахара вважається мозкова мальформация.

В результаті після безлічі досліджень було прийнято спільну думку, що структурні порушення півкуль мозку провокатори захворювання.
Патофізіологія
Синдроми Отахара, і дуже тісно пов'язані між собою, прийнято, що ці синдроми показують вікові реакції мозку в різний період його формування.
з цим видом епілепсії доводилося стикатися ще хворим з полушарнимі або вогнищевими мальформаціями, породжують важкі часткові напади, які передують, але в основному слідують відразу за синдромом Отахара.
Клінічні прояви
У 2002 році Aicardi і Ohtahara представили наступні головні особливості захворювання Отахара:
- хворіють , як правило, немовлята відразу після народження або в 10-денному віці;
- різні типи нападів, головний — збудливий спазм, напади з надмірним напруженням м'язів можуть проявлятися як вдень, так і вночі;
- важке уповільнення в психотропном формуванні, часто закінчується смертю ще в дитячому віці;
- можлива трансформація хвороби в інші синдрому;
- в основному,всі випадки пов'язані з порушеннями мозку.
Для синдрому Отахара властиво прогресуюче погіршення стану здоров'я з почастішанням кількості нападів, видиме уповільнення в психомоторному формуванні організму. В основному, діти з таким діагнозом лишаються інвалідами.
Для захворювання характерні різні види нападів. Як правило, збудливі спазми, які можуть поширюватися по всьому організму, симетричні і латералізованние щодо півкуль мозку. Середня протяжність нападу в середньому 10 секунд, з інтервалом 10-15 секунд. Також є невелика ймовірність в прояві інших нападів.
Через захворювання немовлята мало проявляють активність, виявляється . У синдром Веста хвороба може трансформувати на 2-6 місяці життя дітей, за підсумками досліджень відсоток переходу становить близько 75%. Надалі захворювання має всі шанси перерости в синдром Леннокса-Гасто.
Діагностичні процедури
Нейровізуалізація — сукупність способів, за допомогою яких можна візуалізувати будова, функції та біохімічні властивості мозку. Дані методи необхідні для з'ясування причин і призначення лікувальних терапій. Як правило, при цьому виявляють значні відхилення від норми і мальформації.
 Якщо ж результати нейровізуалізації в нормі, проводитися метаболічний скринінг.
Якщо ж результати нейровізуалізації в нормі, проводитися метаболічний скринінг.
При перших симптомах захворювання проводиться інтеріктальном електроенцефалографія, яка представляє собою патерн «спалах-пригнічення» з високою амплітудою нападоподібними розрядами, які відокремлені один від одного плоскої кривої з тривалістю близько 18 секунд.
патерн «спалах-пригнічення» може бути несиметричний або переважати в одному з півкуль мозку, а також погіршуватися в момент сну.
Якщо на 3-5 місяці патерн «спалах-пригнічення» місцями заміщається гіпсарітмія (аномальна хаотична активність), то це говорить про інше захворюванні — синдромі Веста.
Якщо повільна спайк-хвильова активність, то це властиво для синдрому Леннокса-Гасто. У всіх інших ситуаціях переростає у важку з високою активністю нервових клітин в одному з півкуль.
Так як головна причина захворювання — порушення головного мозку, то необхідно провести перевірку способом нейровізуалізації. Всі структурні зміни можна побачити за допомогою і КТ.
При нормальних результатах нейровізуалізації призначаються діагностика метаболізму. Деякі розлади в процесі обміну речовин оргазму можуть привести до поразок півкуль головного мозку.
Ефективність терапії практично нульова
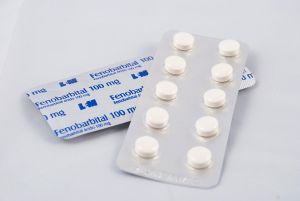 Протиепілептичний лікарський засіб , також відоме під маркою Люмінал, здатне знизити кількість нападів, але антиконвульсанти не в змозі припинити уповільнення психомоторного формування.
Протиепілептичний лікарський засіб , також відоме під маркою Люмінал, здатне знизити кількість нападів, але антиконвульсанти не в змозі припинити уповільнення психомоторного формування.
Ні в одному з відомих випадків не було позитивної реакції на лікування адренокортикотропним гормоном і антагоністів кальцію. У 2001 році Fusco продемонстрував сприятливу терапію вітаміном B6.
Ohno представив ситуацію з хорошим результатом на спосіб лікування зонізамід. У варіантах з гемімегаленцефаліей або кортикальной дисплазією можуть допомогти нейрохірургічні втручання.
На жаль, в даний час не існує дієвого медикаментозного лікування хвороби, половина пацієнтів помирає ще в дитячому віці від декількох тижнів до місяця, у решти розвивається стійкий неврологічний і психологічний недолік.
Часто напади при синдромі Отахара не підлягають ремонту і не піддаються лікуванню антиепілептичних лікарськими засобами. Згодом можлива трансформація в інші хвороби.
Якщо перехід не був здійснений, то психомоторне розвиток буде краще. Однак прогноз несприятливий, в більшості випадків спостерігався летальний результат.