
Мукополісахарідоз відноситься до групи спадкових патологій сполучної тканини, які обумовлюються порушеннями в обміні глікозаміногліканів (кислихмукополісахаридів) і є результатом обумовленої генетично неповноцінності ферментів, які беруть участь в їх розщепленні . Дана патологія успадковується по аутосомно-рецесивним типом.
При мукополісахарідозов відбувається ураження системи лізосомних ферментів, які беруть участь в катаболизме гликозаминогликанов. Через брак ферментів глікозаміноглікани у великій кількості скупчуються в тканинах і органах, тому мукополісахарідоз прийнято відносити до захворювань накопичення. В кінцевому підсумку порушується функціональний стан різних систем і органів, а так як гликозамин присутні в складі сполучних тканин, то одним з головних клінічних проявів мукополисахаридоза виступає системне ураження скелета, а також затримка у фізичному розвитку, особливо це характерно для типів IS і VI.
 Відповідно до сучасної класифікації на сьогоднішній день можна виділити всього вісім типів даного захворювання в залежності від ферментативного дефекту. П'ять з них є основними, а решту три відносяться до інших типів мукополисахаридоза. У літературі можна зустріти медичний термін «гаргоілізм», який введений в медицину в 1936-му році англійським лікарем Еллісом, цей термін об'єднує мукополісахарідоз I-S і II типів. Далі в статті ми більш детально розглянемо всі існуючі типи цього захворювання і характерні для них клінічні прояви.
Відповідно до сучасної класифікації на сьогоднішній день можна виділити всього вісім типів даного захворювання в залежності від ферментативного дефекту. П'ять з них є основними, а решту три відносяться до інших типів мукополисахаридоза. У літературі можна зустріти медичний термін «гаргоілізм», який введений в медицину в 1936-му році англійським лікарем Еллісом, цей термін об'єднує мукополісахарідоз I-S і II типів. Далі в статті ми більш детально розглянемо всі існуючі типи цього захворювання і характерні для них клінічні прояви.
Мукополісахарідоз типу IH
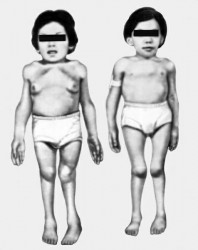
Мукополісахарідоз типу IH, інакше відомий як синдром Гурлера, зустрічається у одного новонародженого дитини з 20-25 тисяч. Симптоматика захворювання з'являється протягом першого року життя дитини, а формування повної клінічної картини відбувається до одного-двох років життя.
При даному типі мукополисахаридоза характерною особливістю є деформація черепа в області човнового кіля (скароцефалія), а також типові грубі риси обличчя. Через занадто великих аденоїдів, а також дефектів розвитку області особи, зокрема носа, хворі зазвичай дихають через рот. Можна відзначити наявність прогресуючих деформацій верхніх і нижніх кінцівок, а також інших частин скелета, значне відставання в рості.
Хребет хворих мукополісахаридозом викривлений, тому в сидячому положенні характерний так званий «синдром котячої спини». Присутній високе розташування лопаток, вкорочення шиї і вистояніе нижніх ребер. Кисті рук дуже широкі, за зовнішнім виглядом нагадують пазури. З плином часу у пацієнтів з даною патологією відбувається формування суглобової контрактури. Спершу, уражаються плечові і ліктьові суглоби, а потім — суглоби коліна, гомілковостопні і тазостегнові. Унаслідок обмеженою рухливості з'являється своєрідна хода — навшпиньки і з напівзігнутими ногами.
Тони серця при цій хворобі приглушаются, а межі розширюються. При проведенні аскультации можна виявити наявність систолических шумів на електрокардіограмі, а також дифузне ураження міокарда. Передня черевна стінка хворих мукополісахаридозом ослаблена, живіт збільшується в розмірах, досить часто присутні гидроцеле, пахові і пупкові грижі. Також збільшені селезінка та печінка. Типовими є порушення слуху і зору. Зустрічається збільшення і помутніння рогівки, пігментна дистрофія сітківки, атрофія зорових нервів, глаукома, а також застійні процеси в області очного дна. Для пацієнтів, які страждають від мукополисахаридоза цього типу, характерна розумова відсталість, яка з часом прогресує. Можливі проблеми з координацією рухів, паралічі і парези. Типовий посилений ріст Пушкова волосся.
При проведенні рентгенологічного дослідження хребта хворих мукополісахаридозом визначаються характерні деформації хребців. Вони мають кубовидной формою, контури їх закруглені, в перехідному відділі можна виявити углообразний кіфоз, скошенность передневерхніх кутів, вкорочення і потовщення відростків. На рентгені грудної клітини видно витончення задніх і потовщення передніх відділів ребер, які супроводжуються їх шаблевидної або ж лопатоподібної деформацією, деформація і укорочення ключиць, зміщення і зменшення в розмірах головок плечових кісток.
На рентгенограмі тазу підтверджується звуження тазового кільця, а також зменшення головок стегнових кісток і скошенность вертлюжної западин. При проведенні рентгена трубчастих кісток можна виявити розширення кістковомозкового каналу і витончення кортикального шару. Рентгенограма кісток кисті підтверджує недорозвинення фаланг нігтів, розширення і скорочення п'ясткових кісток, середніх і проксимальних фаланг. Рентген черепа визначає недорозвинення кісток лицьового черепа, Макроцефалія і краніостеноз.
Мукополісахарідоз типу IS
Мукополісахарідоз типу IS, інакше званий хворобою Шейе (в честь американського офтальмолога Шейе, який в 1962-му році і описав цю патологію) представляє з себе більш пізній, щодо сприятливо протікає варіант мукополисахаридоза IH (або синдрому Гурлера).
До трьох-шести років розвиток дитини відповідає нормальному. Першим симптомом мукополисахаридоза виступає поява згинальних контрактур пальців рук. Потім спостерігаються обмеження розгинання плечових іпроменезап'ясткових суглобів. У більшості випадків контрактури на ногах виражені незначно. Формування повної клінічної картини хвороби відбувається до початку статевого дозрівання дитини.
Пацієнти з цим типом мукополисахаридоза відрізняються кремезний, невисокого зросту, сильно розвиненою мускулатурою і грубими рисами обличчя. Можна відзначити підвищений оволосіння (або гіпертрихоз). Найчастіше розвиваються пупкові або пахові грижі. Шкірний покрив на пальцях хворого потовщений і сильно натягнутий. Внаслідок передавливания серединного нерва може виникати синдром зап'ястного каналу, який супроводжується парестезіями в області третього-четвертого пальців і атрофією м'язів в області тенара. У багатьох пацієнтів з мукополісахаридозом можна виявити недостатність клапанів аорти, аортальний стеноз, пігментну дистрофію сітківки, а також помутніння рогівки. Нехарактерно зниження інтелекту, збільшення печінки або селезінки. При проведенні рентгенологічного дослідження видно чітка картина, така ж, як і в разі мукополисахаридоза типу IH, але при типі IS патологічні зміни виражені не так сильно.
Мукополісахарідоз типу II
Мукополісахарідоз типу II ( інша його назва синдром Хантера) виявляється у пацієнтів-хлопчиків і в більшості випадків розвивається на другому-третьому роках життя. Також як і у випадку мукополисахаридоза типу IH виявляється скафоцефалія, низький голос, огрубіння рис обличчя, труднощі з диханням внаслідок деформації лицьового скелета. Але характерний для хвороби типу IH кіфоз, як правило, відсутня, симптому «котячої спини» також немає. Хворі цією формою мукополисахаридоза досить часто страждають від різних респіраторних інфекційних захворювань (таких як пневмонії, трахеїти, бронхіти). З плином часу характерна поява порушень в координації рухів, відзначається зростання агресивності. Настрій і емоційний стан хворого дуже нестійкі, схильні так і різко змінюватися.
Крім цього, при даному типі мукополисахаридоза можна спостерігати незначні збільшення в розмірах печінки та селезінки, туговухість, посилюється з часом, з'являються вузлики на спині, характерно несильно зниження інтелекту. Надалі можливо помутніння рогівки. Рентгенологічне дослідження виявляє те ж саме, що і при хворобі типу I-S. При даній формі можливі два варіанти розвитку патології: варіант А (або сприятливий) і варіант Б (несприятливий). При сприятливому варіанті сімпоматіка хвороби виражена незначно, іноді може з'явитися несильний розумова відсталість, люди з мукополісахаридозом можуть дожити до 30-ти років і навіть більше. У разі несприятливого варіанта типові яскраві клінічні прояви патології, присутні сильні порушення в розумовому розвитку хворого. Летальний результат у другому варіанті настає в підлітковому віці.
Мукополісахарідоз типу III
Мукополісахарідоз типу III або по іншому хвороба Санфіліппо (була описана американським педіатром на ім'я Санфіліппо в 1963-му році) з'являється у одного новонародженого з 100 000-200 00.У перші роки життя розвиток дитини відповідає її віку, іноді можна спостерігати незграбну ходу і труднощі при ковтанні. У віці від 3-х до 5-ти років дитина з мукополісахаридозом раптово стає апатичним, починають проявлятися відставання в розвитку. Характерно поява порушень мови, огрубляются риси обличчя, можливо нетримання калу і сечі. З ходом часу спостерігається прогресування розумової відсталості, яка займає чільне місце в клінічній картині цієї форми захворювання.
Спільно з порушеннями в інтелекті типово збільшення селезінки і печінки, посилений ріст волосся на тілі, наявність контрактур і затримки в рості. Нехарактерними при даній формі мукополисахаридоза є порушення з боку серцево-судинної системи і органів зору. На рентгені не визначаються значні зміни, картина може бути аналогічною мукополісахарідозов типу I-S, але не так сильно виражена. Здебільшого пацієнти з мукополісахаридозом 3-го типу вмирають у віці від 10-ти до 20-ти років через ускладнень від перенесених інфекцій.
Мукополісахарідоз типу IV
Мукополісахарідоз типу IV, по іншому відомий, як хвороба Морков (дана патологія була описана педіатром з Уругвайя Морков і 1929-му році) зустрічається у одного новонародженого з 40 000. У віці від року до трьох років розвиток дітей відповідає нормі. Надалі майбутньому спостерігається сильне відставання в рості, вкорочення тулуба і шиї, з'являються контрактури, , вальгусна деформація нижніх і верхніх кінцівок, кіфоз, різні деформації грудної клітки, зменшується м'язова сила, потовщується шкірний покрив, і огрубляются риси особи. При цій формі мукополисахаридоза типово розвиток пупкових і пахових гриж, приглухуватості і виникнення дистрофії рогівки. Інтелектуально розвиток не страждає.
При проведенні рентгенологічного дослідження виявляється сколіоз, кіфоз, сплощення і розширення тіл хребців. На рентгені таза, а також нижніх і верхніх кінцівок добре видно множинні деформації, сплощення головок стегнових кісток, нерівні контури, вкорочення кісток передпліччя, деформації стопи. У середньому тривалість життя пацієнтів з мукополісахаридозом не досягає двадцяти років. Летальний результат за цієї форми хвороби настає внаслідок супутніх патологій, які ускладнює серцево-легенева недостатність.
Інші типи мукополисахаридоза
Крім описаних вище основних типів даного захворювання можна виділити також три додаткових а саме: мукополісахарідоз VI типу, VII типу і VIII типу. Далі ми розглянемо їх більш детально:
1.Мукополісахарідоз VI типу (інакше званий синдромом Марото-Ламі або хворобою Морото-Ламі) — перша симптоматика даної форми починає з'являтися у дітей у віці після 2-х років. Типовими особливостями є відставання в рості, огрубіння рис обличчя (таке ж як і при синдромі Гулер, але не так сильно виражений), маленькі розміри верхньої щелепи, бочкообразная грудної клітки, надто коротка шия, укорочені ключиці.
Можна отметітьпоявленіе згинальних контрактур суглобів рук (хворі не в змозі самостійно піднімати руки вгору), з ходом часу виникають контрактури і в суглобах нижніх кінцівок, характерні порушення ходи. Досить часто сюди ж приєднуються часті гострі респіраторні інфекції. Можна нерідко виявити і грижі, а також гепатоспленомегалію. У ряді випадків може розвинутися гідроцефалія, а також спастичні паралічі. Інтелектуальні здібності хворого при даній формі мукополисахаридоза не страждають.
За ступенем вираженості симптоматики мукополисахаридоза захворювання Марото-Ламі можна поділити на типову класичну форму (форму А) або більш м'яку (форму В), при якій клінічні прояви виражені не так сильно.
2.Мукополісахарідоз VII типу — по іншому називається синдромом Слая. Ця форма мукополисахаридоза була описана Слаем в 1973-му році. Симптоми даного типу хвороби дуже схожі з синдромом Санфіліппо (або мукополісахаридозом третього типу). Точно діагностувати цю патологію можна виключно після проведення біохімічного дослідження.
3.Мукополісахарідоз типу VIII (інша назва синдром Ді Ферранте). Патологія була описана медиком Ді Ферранте в 1978-му році. Клінічні симптоми цієї форми дуже схожі на мукополісахарідоз четвертого типу (або синдром Морков). Єдина відмінна риса — це те, що в разі мукополисахаридоза восьмого типу проявляється ще й затримка інтелектуального, а також психомоторного розвитку.
Який лікар лікує мукополісахарідоз?
У разі підозри на дану патологію, лікування має спрямовуватися на ліквідацію симптомів захворювання. У цьому випадку пацієнт в більшості випадків спостерігається одночасно у ряду фахівців, а саме:
- у ортопеда (який займається корекцією порушень опорно-рухового апарату і скелета);
- хірурга (в якщо буде потреба він усуває грижі);
- педіатра (пацієнт стоїть на контролі у цього лікаря через часто розвиваються респіраторних захворювань);
- кардіолога (який займається лікуванням серцево-судинної недостатності хворих);
- отоларинголога (контролює проблеми зі слухом, лікує хронічні синусити і отити);
- офтальмолога (стежить за гостротою зору у пацієнтів з мукополісахаридозом);
- невропатолога (розбирається з неврологічними порушеннями).
Так як мукополісахарідоз є, перш за все, генетичної патологією, то його лікуванням також займається лікар-генетик.
На завершення даної статті потрібно сказати пару слів щодо прогнозу даного захворювання. При всіх типах мукополисахаридоза прогноз несприятливий, так як з віком все більше наростають зміни скелета, а також порушуються функції різних органів і систем організму, тому летальний результат може наступити в будь-якому віці хворого.