
Лейкодистрофії (прогресуючі склерози мозку) — група спадкових захворювань нервової системи, обумовлених порушеннями метаболізму ліпідів мієліну.
Успадковуються лейкодистрофии по аутосомно-рецесивним типом , за винятком суданофільной лейкодистрофии, для якої характерно рецессивное, зчеплене з підлогою, успадкування.
Породжений генетичними факторами ензимний дефект призводить до порушень перебігу мієлінізації. Вироблений організмом мієлін розпадається. У свою чергу, його часткова або повна відсутність веде до поразки білої речовини, породжуючи деменцію. Певні форми даної хвороби більш характерні для пацієнтів чоловічої статі.
Як правило, перші прояви лейкодистрофии головного мозку наздоганяють хворого в дитинстві (ще до вступу до школи). Помітно рідше це відбувається в підлітковому і юнацькому віці. Дана особливість характерна для всієї групи захворювання. Одним з перших симптомів хвороби є порушення рухової функції. Це може бути параліч або парез, рідше виражений гиперкинез. Відзначаються розлади в координації рухів, дизартрія. Трохи пізніше, внаслідок прогресуючого перебігу, захворювання може призвести до судом в різних частинах тіла і поразки зорових нервів.
У великому списку симптомів значаться прогресуючі мозочкові, пірамідальні і екстрапірамідальні розлади, зниження якості та гостроти зору, а також слуху, наростаюча деменція , епілептиформні припадки. Вкрай рідко пацієнти відзначають також порушення чутливості. У міру прогресування захворювання, клінічна картина доповнюється симптомами деградації психіки хворого, спастичними парезами. Термінальна стадія, як правило, характеризується децеребрационной ригидностью.
При проведенні морфологічних досліджень, фахівці виявляють в тканини мозочка і мозку хворого локалізації розпаду мієліну, найчастіше симетричні і слабо обмежені, дифузні. Виражено накопичення продуктів розкладання мієліну в тканинах різних органів, в т.ч. і мозку.
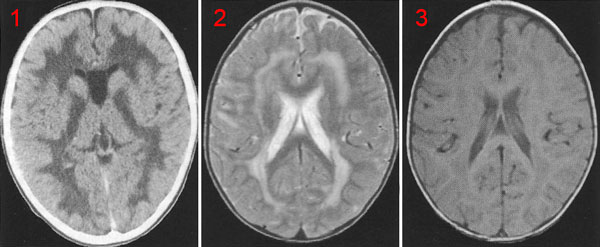
Діагностика захворювання проводиться виходячи зі скарг пацієнта, виявлених клінічних симптомів. Важливу роль в постановці діагнозу грає інформація, отримана після проведення біохімічного аналізу, КТ та МРТ головного мозку. Методи лікування також індивідуальні і залежать від поставленого діагнозу, стану організму і стадії захворювання. Рішення про застосування того чи іншого типу терапії фахівець приймає на підставі даних, отриманих при проведенні діагностичних заходів. До лейкодистрофії відносяться:
- метахроматическая лейкодистрофия (хвороба Шольца),
- суданофільная лейкодистрофия Пеліцеуса- Мерцбахера,
- глобоідной-клітинна лейкодистрофия (хвороба Краббе),
- окремі рідкісні і комбіновані форми.
метахроматичні лейкодистрофия
Вперше дане захворювання було відзначено А. Альцгеймером на початку минулого століття (а точніше, в 1910 році). Більш детально його вивчили в 1925 році під керівництвом лікаря-невропатолога Шольца. Ці та пізніші наукові роботи дозволили медицині з'ясувати, що причиною захворювання є спадкове зміна гена арилсульфатазу А, розташованого в межах 22 хромосоми. Дане відхилення веде до порушеного процесу перетворення сульфатідов в галактоцереброзід. В результаті надмірна кількість сульфатідов 'дрейфують' в організмі людини, накопичуючись в межах печінки і нирок, рідше зачіпаючи легені та серце, а також селезінку, шлунково-кишкового тракту, шкіру і кістки. Як правило, функціональність органів при цьому не падає. Сульфатиди завдають шкоди жовчному міхурі, викликаючи його патологічне функціонування, а також шкодять працездатності ЦНС і периферичної нервової системи. Накопичуючись в білій речовині, ці елементи викликають демієлінізацію мієлінової оболонки нервів периферії.
Ці зміни в організмі позначаються в групі симптомів, що визначають різні стадії і форми захворювання. метахроматичні лейкодистрофия в своїй течії різниться як пізня інфантильна (в чотирьох стадіях), ювенільний і доросла форми. Початок хвороби проявляється в м'язової слабкості і гіпотонії кінцівок, порушенням сухожильних рефлексів, утрудненнями в ходьбі. Надалі дитина демонструє відсталість розвитку (фізичного і інтелектуального), обмежена рухомість, тяжкі порушення психічного здоров'я. У більшості випадків хворий вмирає в ранньому віці, рідше летальний результат наступає в період ювенільної форми (після 3 і до 10 років), ще рідше — дорослої форми (в період від 40 до 50 років).
Діагностика і лікування в кожному випадку індивідуальні і залежать від показань, отриманих від пацієнта, його оточення і на підставі діагностичних заходів. В цілому ж, лікування, здатне запобігти летальному результату, ще не знайдено. Для полегшення стану пацієнта робляться спроби нормалізувати синтез сульфатідов.
лейкодистрофії Краббе
Дана форма лейкодістрфіі виражається в дегенеративном ураженні нервової системи людини прогресуючого характеру. Дефект, що приводить до розвитку хвороби, успадковується по аутосомно-рецесивним типом і призводить до зниження концентрації і активності галактозілцерамід-b-галактозидази. В результаті процес розщеплення галактоцереброзіда до цераміду і галактози порушується або сповільнюється, що призводить до підвищеної концентрації галактоцереброзіда в органах, а також до нестачі галактози і цераміду.
Вільний галактоцереброзід накопичується в білій речовині головного мозку, а також у печінці, нирках і селезінці, в лейкоцитах і фібробластах. Значне (від 10 до 100 разів) підвищення концентрації даної речовини призводить до деміелізаціі, внаслідок чого периферичні нерви виявляються схильні до аксональной дегенерації.
лейкодистрофії Краббе , як правило, проявляється після досягнення 4 місяці життя. У цьому час дитина сильно порушимо, і проявляє м'язову гіпертонію. Постійно розігнуті кінцівки і стиснуті кулаки, а також подальша відсталість в психомоторному розвитку видають хвороба. В подальшому може спостерігатися спастичний тетрапарез, порушення сухожильних рефлексів, характерна рухова реакція в разі виникнення слухових подразників. При огляді очного дна може бути відзначена атрофія зорового нерва. У рідкісних випадках можлива периферична нейропатія. Як правило, летальний результат наступає в період від 7 місяців до 3 років.
Діагностика і прийняття рішення щодо можливого методу полегшення стану хворого відбувається на підставі біохімічного аналізу цереброспінальної рідини. Специфічні лікування даного захворювання відсутня. Нерідко діагностика проводиться пренатально.
Патологічна анатомія лейкодистрофії
Основними змінами, виявленими при морфологічному дослідженні головного мозку хворих, які померли від лейкодистрофии, є набряк і набухання мозкових оболонок, внутрішня гідроцефалія. Кора головного мозку різко стоншена, біла речовина ущільнено, звивини мозку вузькі, борозни глибокі. Виражені дифузний глиоз і склероз головного мозку, перицелюлярний іпериваскулярний набряк. Особливо страждає біла речовина, ураження нервових клітин, очевидно, має вторинний характер. При метахроматіческая лейкодистрофии виявлені метахроматичні речовини, що представляють липоиди і »групи сульфатідов, в ділянках демієлінізації, нервових клітинах і в деяких внутрішніх органах. При глобоідной-клітинної формі лейкодистрофии в головному мозку виявляються глобоїдні клітини. У великій кількості їх виявляють в ділянках значної демиелинизации. Походження клітин адвентіціальние-гістіоцитарні, вони грають роль фагоцитів. У хворих з суданофільной лейкодистрофією Пеліцеус-Мерцбахера поразку мієліну в головному мозку має плямистий характер, що проявляється чергуванням збережених і деміелінізірованних ділянок. Вогнища демієлінізації виконані суданофільнимі зернистими кулями ,, багато з клітин збільшені в розмірах, потворної форми, мають два або більше ядер. Близько вогнищ демієлінізації спостерігається дифузна проліферація волокнистої астроглії. Навколо судин у вигляді муфт значна кількість плазматичних клітин. Вогнища демієлінізації розташовані дифузно в великих півкулях, стовбурі мозку і мозочку. Переважна локалізація ділянок демієлінізації навколо судин і наявність суданофільних зернистих куль представляють характерні морфологічні риси цієї форми лейкодистрофии. Поряд з цим є виражена реакція гліальних тканини і серйозні дистрофічні зміни нервових клітин кори великих півкуль і підкіркових вузлів.
Патогенез лейкодистрофії
При лейкодистрофії відбувається накопичення певних ліпідів в різних органах і тканинах. Так, при метахроматіческая лейкодистрофии в білій речовині мозку накопичуються сульфатиди і знижений рівень фосфоліпідів, холестерину, протеоліпіди, в сірій речовині зміст ліпідів істотно не змінено. При глобоідной-клітинної лейкодистрофии (хвороба Краббе) в білій речовині головного і спинного мозку відзначаються виражена демиелинизация, накопичення цсреброзідов при зниженні фракцій інших ліпідів. При суданофільной лейкодистрофии виявлено зменшення всіх ліпідних фракцій, крім сфінгоміеліна [Blachwood, Cumings, 1954].
Клініка лейкодистрофії
Провідними симптомами в клінічній картині лейкодистрофії є неврологічні порушення, що відрізняються прогредієнтність. Поява клінічних симптомів відзначається переважно в перші роки життя. В окремих випадках дебют захворювання може відзначатися в шкільному і молодому віці (ювенільний форма). Як ранніх ознак часто виявляються судомні напади, гіперкінези, спастичні парези кінцівок. У міру розвитку захворювання можуть приєднуватися інші неврологічні симптоми: косоокість, тремор голови, розлади координації, зміна тонусу м'язів, зниження інтелекту, втрата набутих мовних функцій, вегетативні порушення. На очному дні з великою постійністю виявляється первинна атрофія сосків зорових нервів. У спинномозковій рідині виявляється білково-клітинна дисоціація.
Клініка метахроматіческая лейкодистрофии
Типовими ознаками метахроматіческая форми лейкодистрофии є симптоми, які вказують на дифузне ураження нервової системи. Особливо часто виявляються спастичні парези та паралічі, атаксична синдром, псевдобульбарние симптоми, прогресуюча деменція, зниження зору і слуху, поліморфні судомні напади. Характерні вегетативні розлади: набряки, зниження харчування, гіпергідроз або сухість шкіри, гіперсалівація. Перебіг захворювання носить прогресуючий характер. У заключній стадії метахроматіческая лейкодистрофии виявляються стану децеребрационной ригідності, почастішання судомних нападів, гіпертермічні кризи. Летальний результат наступає в результаті пневмонії, серцевої і дихальної недостатності. При лабораторних дослідженнях відзначені дифузнізміни на ЕЕГ, білково-клітинна дисоціація в спинномозковій рідині, часто без підвищення лікворного тиску — гідроцефалія ex vacuo, атрофія зорових нервів. Залежно від характеру клінічної симптоматики метахроматіческая лейкодистрофією необхідно диференціювати з дитячим церебральним паралічем, епілепсією, спінальної аміотрофією, енцефалітом. Уточненню діагнозу сприяють біохімічні дослідження, які виявляють специфічні зміни в обміні сульфатідов — позитивний тест Аустін з сечею — поява золотисто-коричневих тілець, видимих під мікроскопом при фарбуванні осаду сечі толуїдиновим синім, тест Аустін є специфічним для метахроматіческая форми лейкодистрофии, а також зміна ліпідів в крові і особливо в спинномозковій рідині.
У спинномозковій рідині у хворих метахроматіческая лейкодистрофією виявляється підвищення рівня цереброзидів (за рахунок сульфатідов) в середньому до 0,53 мг% (в нормі становить 0,24 мг%).
Оскільки сульфатиди є важливим компонентом мієліну, їх зміну можна розглядати як результат порушення метаболізму ліпідів мієліну — дісміелінізацію.
Клініка глобоідной-клітинної лейкодистрофії (лейкодистрофии Краббе)
Найбільш злоякісний перебіг захворювання відзначається у хворих з глобоідной-клітинної лейкодистрофією. Захворювання починається на першому році життя, частіше до 6 міс. У хворих розвиваються судомні напади, які мають клонико-тонічний характер. Відзначаються також малі припадки. У міру розвитку хвороби судомні напади стають частішими і тривалими. Діти відстають у фізичному і психічному розвитку, втрачають раніше набуті навички, перестають впізнавати батьків. Відзначається в початкових стадіях хвороби гіпотонія м'язів змінюється їх гіпертонією, в розгорнутій стадії глобоідной-клітинної лейкодистрофии у всіх хворих виявляються спастичность і в подальшому — стан децеребрационной ригідності. Нерідко виникають гіпертермічні кризи — температура тіла піднімається до 40-41 ° С і утримується протягом кількох днів. Цікаві деякі зовнішні риси хворих: велика голова, світле волосся, коротка шия, відзначаються також різноманітні диспластические ознаки. На очному дні виявляється часткова або повна атрофія зорових нервів. Поступово у хворих розвивається кахексія. Характерні також гідроцефалія, бульбарні розлади, органічна деменція. У спинномозковій рідині наростає білково-клітинна дисоціація. У плазмі крові хворих виявляється різке підвищення рівня вільного холестерину, що перевищує нормальні показники в 3.5-9 разів. Реєструється також помірне підвищення окремих фракцій фосфоліпідів. Дослідження ліпідів в еритроцитах периферичної крові виявляє зниження кількості сфінгоміеліна.
Клініка суданофільной лейкодистрофии Пеліцеус — Мерцбахера
Своєрідна клінічна картина спостерігається у хворих з суданофільной лейкодистрофією Пеліцеус — Мерцбахера. Хворіють хлопчики, іноді є рідними або двоюрідними братами (матері — рідні сестри). Ранніми неврологічними симптомами є ністагм, тремор голови, затримка фізичного і психічного розвитку. Ністагм виявляється з народження дитини або в перші місяці життя, за характером є горизонтальним з копіювальний компонентом, іноді відзначається і вертикальний ністагм. Тремор голови спостерігається в спокої, стає особливо вираженим при спробі утримати голову. У всіх хворих є затримка фізичного і психічного розвитку: діти пізно починають утримувати голову, сидіти, стояти, погано говорять і розуміють звернену мову. У окремих хворих поряд із затримкою фізичного і психічного розвитку захворювання дебютує прогресуючої слабкістю в кінцівках, судорожними припадками. В розгорнутій стадії суданофільной лейкодистрофии у всіх хворих виявляються спастичні парези, порушення координації, скандували мова, органічна деменція. Характерні гідроцефалія, косоокість, порушення пігментації, дегенеративні стигми. Перебіг захворювання з віком стає більш повільним, може приймати стаціонарний характер, що представляє певну особливість цієї форми лейкодистрофии. При додаткових дослідженнях виявляють атрофію зорових нервів, у спинномозковій рідині — слабо виражену білково-клітинну дисоціацію. Діагностиці захворювання допомагає встановлення типу успадкування. Основний тип — рецесивний, зчеплений з підлогою, хоча в окремих випадках не виключається і аутосомно-рецесивний тип спадкування [McKusick, 1966]. Біохімічне дослідження ліпідів дозволяє уточнити діагноз. У плазмі крові помірно підвищується вміст сфінгоміеліна і гликолипидов, а також вільного холестерину. В еритроцитах периферичної крові хворих лейкодистрофією Пеліцеус — Мерцбахера зміст сфінгоміеліна зменшено, в той час як рівень Кефалінія і лецитину дещо підвищений.
Збільшується вміст вільного холестерину, рівень гликолипидов в еритроцитах практично не змінюється. Ці дані дозволяють вважати, що для суданофільной лейкодистрофии характерно зниження сфінгоміеліна в еритроцитах і його підвищення в плазмі крові, підвищення вільного холестерину в плазмі і в еритроцитах. Зміни інших ліпідів менш виражені.
Лікування хворих лейкодистрофії проводиться за принципами, ідентичним лікування хворих внутрішньоклітинними Ліпоїдоз.