
 олівопонтоцеребеллярная дегенерації (ОПТСД), як готельний вид захворювання був описаний більш 100 років тому. Протягом всього періоду хвороба вивчалася, розроблялися методи діагностики і лікування.
олівопонтоцеребеллярная дегенерації (ОПТСД), як готельний вид захворювання був описаний більш 100 років тому. Протягом всього періоду хвороба вивчалася, розроблялися методи діагностики і лікування.
Сьогодні відомо, що оливопонтоцеребеллярная дегенерація передається у спадок. Хвороба вражає центральну нервову систему. Локалізація атрофії — мозочок, міст і нижні оливи (відділи головного мозку).
Основним проявом служить зміна в звичному поведінці — відзначаються , . Цей вид захворювання характеризується тривалістю перебігу, але фіксується воно в зрілому віці. У процесі змін, що відбуваються в мозку людини, зазначається , відповідно, порушуються частота проведення сигналів від них в ЦНС.
Також дегенеративно-дистрофічні процеси, які відбуваються на всьому протязі розвитку захворювання, вражають кору мозочка, міст і оливи мозку.
Саме тому спостерігаються характерні зміни в психіці і можливостях людини, включаючи фізичні. У рідкісних випадках під час  діагностики відзначається ураження таких ділянок, як передні роги спинного мозку і його провідні шляхи. Також хвороба може зачіпати каудальні черепно-мозкові нерви (9-12 пар). До порушень, які пов'язані з олівопонтоцеребеллярная дегенерацією, відносять такі захворювання:
діагностики відзначається ураження таких ділянок, як передні роги спинного мозку і його провідні шляхи. Також хвороба може зачіпати каудальні черепно-мозкові нерви (9-12 пар). До порушень, які пов'язані з олівопонтоцеребеллярная дегенерацією, відносять такі захворювання:
- ;
- ;
- .
спадкування захворювання відбувається за рецесивним і домінантним шляху, а також відомі випадки спорадичні успадкування.
Стан хворого може виражатися в втрати орієнтації, провалах в пам'яті, повному або частковому склерозі, порушеннях в ході, скутість рухів, зниженні рухової активності в цілому і тонусі організму, апатії, втрати здатності концентрувати увагу.

Статистичні дані
За наявними відомостями велика частина випадків захворювання відзначається у людей похилого віку — після 60-65 років — 75% випадків.
Однак дегенерація може почати розвиватися і в більш ранньому віці — в період 30-40 років, а в разі успадкування і у дітей у віці 10-11 років (2-3%).
Саме тому проходити обстеження необхідно не тільки по досягненню похилого віку, а й в 20-40 років, а в разі вроджених порушень — постійно протягом усього життя.
Причини розвитку хвороби
Важливою особливістю хвороби є той факт, що точні причини її виникнення і розвитку, якщо не зазначено випадків захворювання в сім'ї, не відомі медицині навіть через століття після першого опису. Відзначаються порушення в локусах генів, що виражаються в збільшенні числа тринуклеотидних повторів.
Серед інших можливих причин, які можуть призвести до появи характерних порушень відзначаються асиметричні атрофічні зміни в білій речовині. Атрофія кори мозочка відзначається на пізніх стадіях захворювання. Для того щоб дізнатися точні причини, необхідно пройти повне дослідження.
Різновиди течії
Виділяють кілька основних видів олівопонтоцеребеллярная дегенерації: 
- тип Менделя;
- тип Фіклера- Вінклера;
- атрофія з ретинальной дегенерацією;
- тип Шута-Хайкмана;
- атрофія з .
Також в окрему категорію можна включити синдром Шая — Дрейджера, який характеризується наступними основними по частоті прояви симптомами:
- вегетативні розлади;
- мозочкові порушення і (різного ступеня вираженості);
- поразку в області базальних гангліїв.
Тип Менделя має аутосомно-домінантний механізм успадкування і наступні прояви:
- повільне і неяскраве розвиток захворювання, що провокує його розвиток і перехід в запущену стадію;
- вік прояву перших симптомів з дитинства і до 60 років;
- ;
- (іноді значне , людина не може виконувати звичайних справ);
- (проявляється від слабкого до сильного);
- тремтіння і посмикування в руках ();
- порушення в процесі ковтання;
- ;
- порушення, пов'язані з рухом очей (рідко).
олівопонтоцеребеллярная дегенерація Фіклера-Вінклера проявляється наступними ознаками:
- успадковується аутосомно-рецесивним способом;
- вік появи симптомів і розвитку захворювання протягом усього життя, починаючи з 20 років;
- атаксія кінцівок.
Якщо діагностується цей вид, то порушення рухової активності не простежується.
Тип з ретинальной дегенерацією має наступні симптоми: 
- аутосомно-домінантний принцип передачі новому поколінню;
- страждають молоді люди і діти;
- ;
- ;
- зміни в гостроті зору (значне падіння), з'являється порушення по причини пігментації сітківки.
Вид Шута-Хайкмана відзначається наступними симптомами та проявами:
- аутосомно-домінантний основний метод наслідування;
- прояви і розвиток в молодому ( 20-30 років) або дитячому віці;
- атаксія;
- характерний для цього виду ознака ;
- порушення ковтання (іноді сильні);
- дефекти мови;
- вібраційні порушення.
Тип з деменцією характеризується наступними ознаками:
- аутосомно-домінантне успадкування;
- прояв і розвиток в середньому віці, але не пізніше 40 років;
- порушення інтелекту (іноді дуже сильне);
- ;
- атаксія.
В цілому симптоматика схожа, але наявні відмінності дають можливість лікарям ставити правильний діагноз і призначати найбільш дієве лікування.
Загальні симптоми
Загальний симптом для всіх видів олівопонтоцеребеллярная дегенерацій — це . На початку у людини відзначається легка нестійкість, потім при швидкій ходьбі пацієнт відчуває дискомфорт, що виражається в незграбних рухах. Додаткові візуальні порушення ходи:
- часті падіння;
- широко розставлені під час ходьби ноги;
- коливання тіла з боку в бік (порушення роботи мозочка);
- нестійке положення під час сидіння на стільці.
Пізні симптоми (хвороба прогресує): 
- великий почерк;
- ;
- тремтіння в кінцівках ();
- ;
- зниження або підвищення рефлексів;
- .
Також до загальної симптоматиці можна віднести:
- порушення ковтання (відзначається в більшості випадків) ;
- .
 Особовий парез не є загальним для всіх видів симптомом, на відміну від нетримання сечі.
Особовий парез не є загальним для всіх видів симптомом, на відміну від нетримання сечі.
Людина майже завжди стає млявим і апатичним, втрачає інтерес до подій навколо нього подій. Нерідко фіксується загальмованість і навіть тупість. Також хвороба супроводжується в різного ступеня вираженості, з'являються депресивні стани, страхи і фобії, які раніше були відсутні. Галюцинації і сплутаність свідомості — характерні симптоми цього захворювання.
Якщо хвороба знаходиться в занедбаному стані, то людина поступово втрачає можливість спочатку ходити, а потім і перестає виконувати найпростіші дії по догляду за собою. Нерідко в цей період у пацієнтів слабшає імунітет, що призводить до розвитку супутніх захворювань.
Діагностичні критерії
Для того щоб діагностувати захворювання, а потім точно дізнатися його тип, потрібно пройти кілька обстежень. Це необхідно для отримання об'єктивної і достовірної інформації. Одним з обстежень є неврологічний статус. Також здійснюється дослідження нейропсихологічних даних.
Всі отримані результати дають лікареві можливість з'ясувати, чи є у пацієнта мозжечковая дегенерація, чи присутній і інші симптоми олівопонтоцеребеллярная дегенерації.
Об'єктивну інформацію можна отримати, пройшовши обстеження за допомогою , а потім і . Для визначення типу захворювання потрібно консультація з фахівцем в області генетики.
Додатково може знадобитися діагностика ДНК. Якщо відзначається значне погіршення зору, то до діагностики додається консультація офтальмолога.
Комплекс заходів що можна зробити?
Спеціального дієвого лікування від цього захворювання не створено. Сьогодні проводяться заходи для підтримки показників в усереднених і наближених до норми значеннях. У більшості випадків проводиться симптоматичне лікування з ухилом в неврологію.
Також наголошується на симптоматику, присутню в момент діагностики.
Основні лікарські препарати — нейрометаболіти, холінолітики, а також загальнозміцнюючі засоби. Для підтримки оптимального м'язового тонусу проводиться лікувальний масаж і заняття ЛФК. Також використовуються такі медикаментозні препарати: 
- глутамінова кислота;
- Церебролізин (уколи);
- ;
- Прозерін.
Активні заняття на свіжому повітрі і прості прогулянки входять в комплекс заходів з лікування хвороби. Якщо відзначаються порушення зору, то в терапію включають відповідні препарати.
Значного поліпшення в стані здоров'я людини відзначатися не буде. Однак проведена терапія дозволить підтримувати показники в межах допустимих тривалий час, так тривалість захворювання в середньому становить 12 років, відомі випадки, коли пацієнти могли боротися з подібними захворюванням протягом 20 років. Основна причина смерті — пневмонія або сепсис, як супутні захворювання.
 Хвороба Мойя — Мойя є рідкісне захворювання, характерне двосторонньої природи прогресуючим перебігом, що призводить до звуження внутрішньочерепних артерій, що в підсумку призводить до збою в кровотоці головного мозку. Саме захворювання в кінцевому підсумку може призвести до розвитку в гострій формі збою в мозковому кровообігу — , недостатнього постачання сірої речовини киснем і живильними речовинами.
Хвороба Мойя — Мойя є рідкісне захворювання, характерне двосторонньої природи прогресуючим перебігом, що призводить до звуження внутрішньочерепних артерій, що в підсумку призводить до збою в кровотоці головного мозку. Саме захворювання в кінцевому підсумку може призвести до розвитку в гострій формі збою в мозковому кровообігу — , недостатнього постачання сірої речовини киснем і живильними речовинами.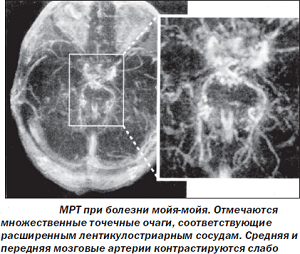
 тепла, ні уколів;
тепла, ні уколів; 
 В щодо призначення консервативного способу лікування, лікарі найчастіше призначають судинну терапію, прописуючи курей прийому таких препаратів як і Інстенон, Серміон, Нифедипин.
В щодо призначення консервативного способу лікування, лікарі найчастіше призначають судинну терапію, прописуючи курей прийому таких препаратів як і Інстенон, Серміон, Нифедипин. Дизартрия, якщо розглядати з суто медичної точки зору — це порушення в роботі мовного апарату.
Дизартрия, якщо розглядати з суто медичної точки зору — це порушення в роботі мовного апарату. 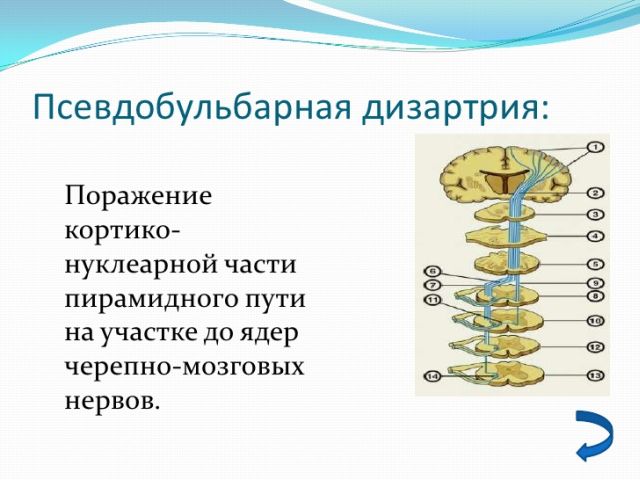
 , своєчасне виявлення якої гарантує повне лікування і можливість дитини в подальшому вести нормальний спосіб життя.
, своєчасне виявлення якої гарантує повне лікування і можливість дитини в подальшому вести нормальний спосіб життя.  Причинами виникнення конкретної форми дизартрії є ураження під'язикового нерва, що визначає чіткість вимови і, як наслідок — подальший прогрес хвороби і ураження кори головного мозку.
Причинами виникнення конкретної форми дизартрії є ураження під'язикового нерва, що визначає чіткість вимови і, як наслідок — подальший прогрес хвороби і ураження кори головного мозку.
 місце сприятливий прогноз, але потрібно визначити — чи немає ще жодних супутніх невралгічних захворювань і порушень.
місце сприятливий прогноз, але потрібно визначити — чи немає ще жодних супутніх невралгічних захворювань і порушень.
 В якості профілактичних заходів для запобігання конкретного захворювання, потрібно звернути найпильнішу увагу на період вагітності і процес пологів.
В якості профілактичних заходів для запобігання конкретного захворювання, потрібно звернути найпильнішу увагу на період вагітності і процес пологів.  Кататонія є цілою групою психопатологічних синдромів, ключовими клінічними проявами яких є рухові порушення.
Кататонія є цілою групою психопатологічних синдромів, ключовими клінічними проявами яких є рухові порушення.  виникають слідом за імпульсивним і безконтрольним поведінкою, мовчанням або .
виникають слідом за імпульсивним і безконтрольним поведінкою, мовчанням або .  вимагає м'язового напруги, не реагує на звуки, однак може реагувати на мова пошепки. Хворий може спонтанно розгальмовує і контактувати в нічний час. При цій формі ступору може спостерігатися марення, і галюцинації.
вимагає м'язового напруги, не реагує на звуки, однак може реагувати на мова пошепки. Хворий може спонтанно розгальмовує і контактувати в нічний час. При цій формі ступору може спостерігатися марення, і галюцинації. При порушенні найбільш проявляються маніакальні риси, які супроводжують пластичність, веселощі, природність рухових дисфункцій, з'являється шізофазія (мовна розірваність.) Характерним є невідповідність між поведінкою і переживаннями хворого. Хворий, проживає в своїх власних насичених і яскравих переживаннях, знаходиться в відмові від навколишнього світу.
При порушенні найбільш проявляються маніакальні риси, які супроводжують пластичність, веселощі, природність рухових дисфункцій, з'являється шізофазія (мовна розірваність.) Характерним є невідповідність між поведінкою і переживаннями хворого. Хворий, проживає в своїх власних насичених і яскравих переживаннях, знаходиться в відмові від навколишнього світу.  Смертельний результат вірогідний через судинної недостатності, яка виникає на тлі набряку головного мозку.
Смертельний результат вірогідний через судинної недостатності, яка виникає на тлі набряку головного мозку.  Основна стратегія лікування кататонических синдромів полягає в призначенні препаратів бензодіазепінового ряду (Лоразепам, ), а також електросудорожна терапія.
Основна стратегія лікування кататонических синдромів полягає в призначенні препаратів бензодіазепінового ряду (Лоразепам, ), а також електросудорожна терапія.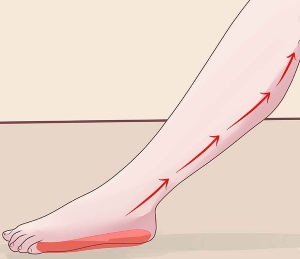 Поразка великогомілкової нерва — це досить серйозне захворювання, яке супроводжується неприємними відчуттями у вигляді сильного болю, крім цього при нейропатії або неврит нижніх кінцівок дуже складно пересуватися і в усьому тілі виникає відчуття сильного дискомфорту.
Поразка великогомілкової нерва — це досить серйозне захворювання, яке супроводжується неприємними відчуттями у вигляді сильного болю, крім цього при нейропатії або неврит нижніх кінцівок дуже складно пересуватися і в усьому тілі виникає відчуття сильного дискомфорту. 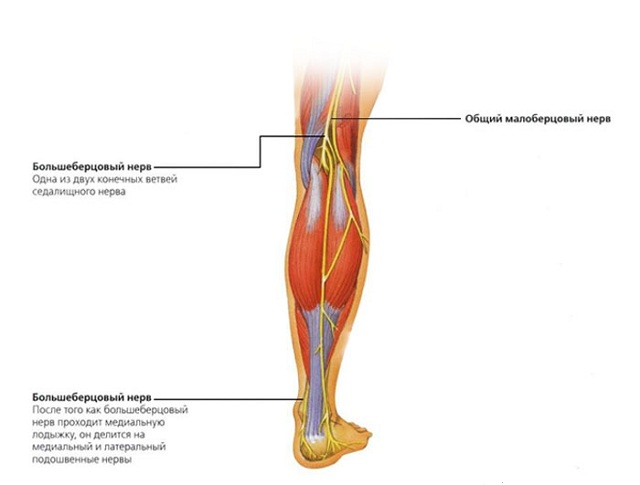
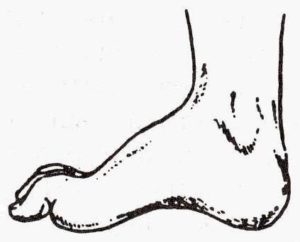 великогомілкової нерва залежить іннервація задньої поверхні гомілки, підошов, підошовної поверхні пальців. При цьому ураженні неможливо зігнути пальці на нозі, стопа також не згинається. Крім цього порушується хода, хворий не може наступати на шкарпетки і пересувається на п'ятах.
великогомілкової нерва залежить іннервація задньої поверхні гомілки, підошов, підошовної поверхні пальців. При цьому ураженні неможливо зігнути пальці на нозі, стопа також не згинається. Крім цього порушується хода, хворий не може наступати на шкарпетки і пересувається на п'ятах. 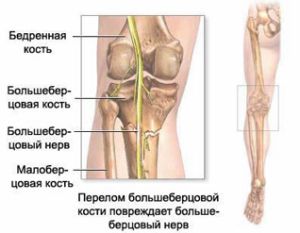 викликає здавлювання нерва і погіршення проведення імпульсів.
викликає здавлювання нерва і погіршення проведення імпульсів. 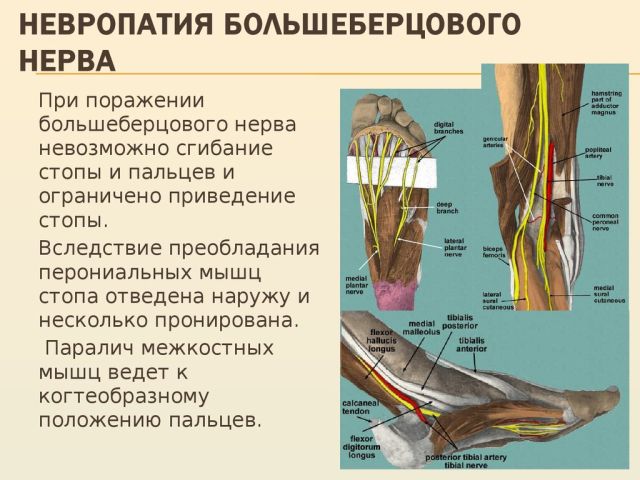
 результаті травматичного пошкодження або ендокринних збоїв, пухлин і так далі.
результаті травматичного пошкодження або ендокринних збоїв, пухлин і так далі. 
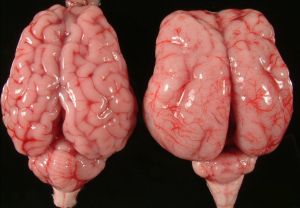 ліссенцефаліі (буквально гладкий мозок) називається вельми велика за обсягом група порушень у розвитку головного мозку, яка характеризуються частковою відсутністю або ж поганим розвитком звивин, розташованих в півкулях головного мозку.
ліссенцефаліі (буквально гладкий мозок) називається вельми велика за обсягом група порушень у розвитку головного мозку, яка характеризуються частковою відсутністю або ж поганим розвитком звивин, розташованих в півкулях головного мозку. 

 .
.  Визначити через що почав формуватися дефект завдання не з легких, так як їх причин бути ціле безліч. Приблизно у 60% людей з ліссенцефаліі помічені мутація в гені LIS1.
Визначити через що почав формуватися дефект завдання не з легких, так як їх причин бути ціле безліч. Приблизно у 60% людей з ліссенцефаліі помічені мутація в гені LIS1. 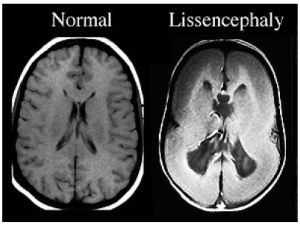
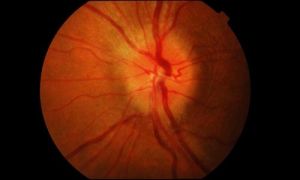 Часто ослаблення або втрата функції органу зору викликані пошкодженням II пари черепних нервів (Nervus opticus), які здійснюють передачу нервових імпульсів від чутливих клітин сітківки в головний мозок.
Часто ослаблення або втрата функції органу зору викликані пошкодженням II пари черепних нервів (Nervus opticus), які здійснюють передачу нервових імпульсів від чутливих клітин сітківки в головний мозок. 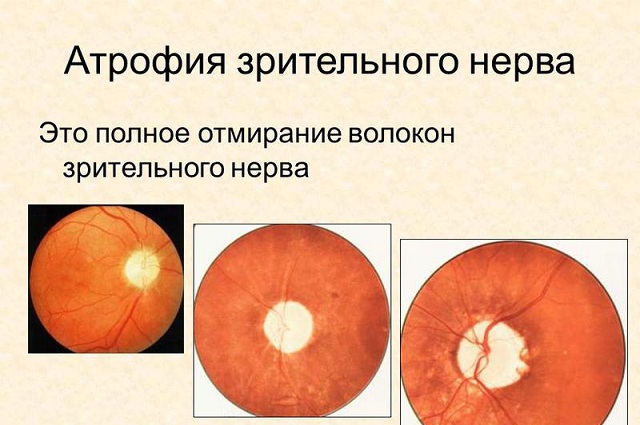
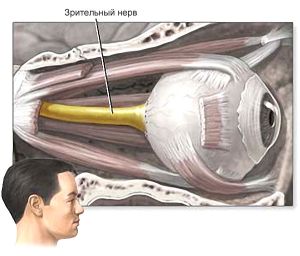
 Головним симптомом дегенеративних процесів в зоровому нерві є погіршення зору, дефекти полів зору різної локалізації, втрата чіткості зорового сприйняття і повноти відчуття кольору.
Головним симптомом дегенеративних процесів в зоровому нерві є погіршення зору, дефекти полів зору різної локалізації, втрата чіткості зорового сприйняття і повноти відчуття кольору. 
 антикоагулянти);
антикоагулянти); 
 Конверсійне розлад (реакція), також зване функціональним неврологічним розладом, являє собою стан, при якому проявляється фізичними почуттями.
Конверсійне розлад (реакція), також зване функціональним неврологічним розладом, являє собою стан, при якому проявляється фізичними почуттями. 




 Підтримка психічного здоров'я та профілактика психічних розладів є набагато менш ясне дію, ніж, наприклад, превенція інфекційних недуг, для профілактики яких досить вакцинації, а терапевтичні заходи засновані на прийомі антибіотиків; в сфері психічних розладів такі дії не передбачені.
Підтримка психічного здоров'я та профілактика психічних розладів є набагато менш ясне дію, ніж, наприклад, превенція інфекційних недуг, для профілактики яких досить вакцинації, а терапевтичні заходи засновані на прийомі антибіотиків; в сфері психічних розладів такі дії не передбачені.  Гіпофіз (пітуітарная заліза або нижній мозковий придаток) невелика залоза внутрішньої секреції, розташована в головному мозку, яка координує роботу всієї ендокринної системи в організмі. Виконує гіпофіз це завдяки виробленню власних гормонів. Її вага приблизно 0,5 г.
Гіпофіз (пітуітарная заліза або нижній мозковий придаток) невелика залоза внутрішньої секреції, розташована в головному мозку, яка координує роботу всієї ендокринної системи в організмі. Виконує гіпофіз це завдяки виробленню власних гормонів. Її вага приблизно 0,5 г. 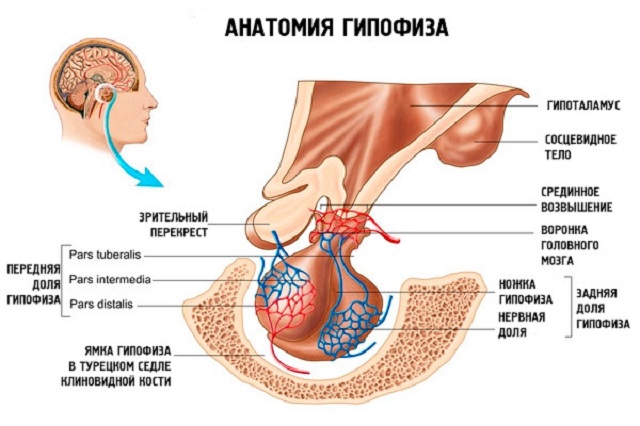
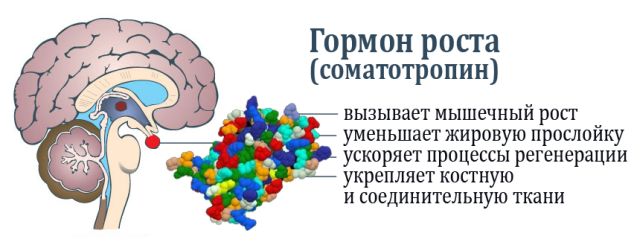
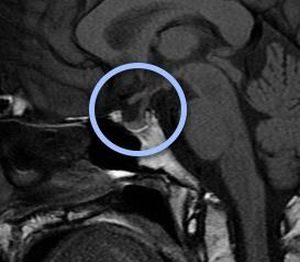
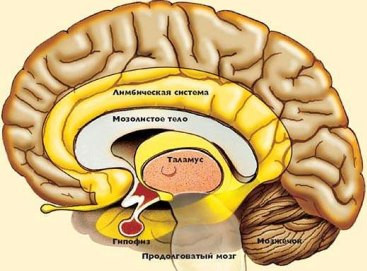 Карман Ратко має ембріональний походження, так як утворюється в процесі закладки і розвитку гіпофіза внутрішньоутробно. Він розташовується між передньою і задньою часткою залози.
Карман Ратко має ембріональний походження, так як утворюється в процесі закладки і розвитку гіпофіза внутрішньоутробно. Він розташовується між передньою і задньою часткою залози.
 Щоб попередити важкі наслідки кісти гіпофіза, необхідно контролювати будь-які зміни гормонального фону. Берегти голову від механічних впливів і . Не менш ніж один раз на рік проходити обстеження у невролога.
Щоб попередити важкі наслідки кісти гіпофіза, необхідно контролювати будь-які зміни гормонального фону. Берегти голову від механічних впливів і . Не менш ніж один раз на рік проходити обстеження у невролога.