Про алергічному характері деяких видів полинейропатий говорять як клінічні спостереження, так і результати експериментальних і морфологічних досліджень. Відома клінічна картина полінейропатії, що розвивається бурхливо і часто завершується одужанням — по алергічного типу — після введення антирабічної вакцини. Були поставлені і експерименти з підшкірним введенням гомо- чи гетерологічних тканин нервів з метою простежити можливість появи алергічної полінейропатії. Виявилося, що при цьому, особливо якщо додати до антигену стимулятор Фрейнда (ліпоподобное з’єднання, парафінова олія і вбиті туберкульозні бактерії), через 15-20 днів простежується поява в сироватці специфічних комплементфіксірующіх і преципитирующих антитіл до речовини зазначених нервів. Одночасно виникає реакція у формі первинно розвивається демієлінізації. Трохи пізніше в сполучної тканини — внутріадвентіціально — в судинах, особливо венулах чутливих гангліїв і задніх корінців виявляються запальні інфільтрати. Вони складаються переважно з моно- і лімфоцитів. Подібними виявилися реакції олігодендроглії мозку і шванновских клітин уражених нервів, а також мезенхимних елементів мозку і структур периневрального бар’єру. Все це дозволило деяким дослідникам стверджувати, що як в експерименті, так і в клініці деміелінізірірующіе енцефаломієліти, енцефаломіелополірадікуліти і алергічні полінейропатії — родинні по етіології і патогенезу стану. Типовим клінічним варіантом такого роду алергічної полінейропатії є хвороба Гійена — Барре. Полінейропатії розвиваються і після введення сироваток, причому нерідко спостерігається зв’язок між появою нейропатії і сироватковоїхвороби — уртикарии, набряками, суглобовими болями. Описано алергічні полінейропатії після ужаливания комахами (бджолами, осами, джмелями). Алергічними є і полінейропатії при колагенозах.
Залежно від характеру антигену, стану сенсибілізації, імунних, аутоімунних, аутоагрессивних преморбідних властивостей організму і чинниками розгортаються різні за характером і течією форми алергічних полинейропатий.
Бічний аміотрофічний склероз (БАС) -важкий прогресуюче захворювання нервової системи, вперше виділений Шарко в 1869 р Незважаючи на більш ніж вікову історію його вивчення, етіологія і багато питань патогенезу залишаються не з’ясованими.
Етіологія бокового аміотрофічного склерозу
Більшість неврологів розглядають БАС як первинний дегенеративний процес, причина виникнення якого не відома. Однак є досить вагомі підстави припускати, що захворювання відноситься до групи так званих повільних інфекцій, викликається вірусом. З іншого боку, показано, що в ряді місць, зокрема на Маріанських островах і острові Гуам в Тихому океані, захворювання, кілька відрізняється по клінічній картині від звичайного БАС-Гуам-тип — носить виражений спадковий характер. В останні роки все більшого поширення набуває уявлення про інфекційне походження захворювання при наявності конституціональної схильності.
При мікроскопічному дослідженні виявляються грубі дегенеративні зміни, що локалізуються переважно в бічних стовпах і передніх рогах спинного мозку. Дегенеративні зміни є також в корі головного мозку, підкіркових утвореннях, мозочку, стовбурі мозку, але інтенсивність їх в цих відділах значно менше. Кількість нервових клітин в передніх рогах спинного мозку різко зменшено, що залишилися клітини патологічно змінені: вони набувають округлу або овальну форму, в них нерідко виявляються явища вакуолизации, нерідко зустрічаються клітини-тіні. Гинуть нервові клітини оточені олигодендроцитов і макрофагами. На місці загиблих клітин розростаються гліальні елементи. Значні зміни виявляються і в білій речовині мозку. Найбільш різко вони виражені в передньо відділах спинного мозку, в області шийного, рідше поперекового потовщення, в меншій мірі в інших відділах спинного та головного мозку. Мієлінова оболонка нервових волокон руйнується, а в багатьох місцях зникають і осьові циліндри. Особливо сильно страждають волокна, що входять до складу пірамідних шляхів.
Клініка бічного аміотрофічного склерозу
Клінічні симптоми БАС обумовлені поразкою як периферичних, так і центральних рухових нейронів.
Основними симптомами захворювання є слабкість і атрофія дрібних м’язів рук, що поширюються потім на м’язи плечового пояса, спини та грудної клітини. В уражених м’язах відзначаються фасцікуляціі, які бувають помітні під шкірою у вигляді швидких неритмічних скорочень окремих рухових одиниць. Їх виникнення може бути спровоковано ударами молоточка. Виникнення фасцикуляций можливо за кілька місяців і навіть років до появи інших симптомів захворювання.
Прогресування захворювання часто супроводжується залученням в процес ядер черепних нервів, розташованих в стовбурі мозку. В першу чергу страждають ядра бульбарной групи нервів.
Порушується мова за типом дизартрії, утруднюється ковтання, з рота весь час виділяється слина. У мові з’являються фібриляції, згодом розвиваються атрофії. Поразка ядер додаткового нерва супроводжується атрофією і слабкістю грудино-ключично-підборіддя і трапецієподібних м’язів. Прогноз в таких випадках стає дуже серйозним, так як поширення патологічного процесу на інші бульбарні структури призводить до летального результату.
Рідше відзначається ураження ядер VII і V пар черепних нервів. Ядра VI, IV і III пари черепних нервів зазвичай в патологічний процес не залучаються. Ураження білої речовини найбільш грубо виражено в бічних і передніх стовпах спинного мозку, а також в області рухової кори і низхідних шляхів. Клінічно це проявляється підвищенням м’язового тонусу і глибоких рефлексів, появою патологічних стопного знаків. Двобічне ураження кортико-бульбарних зв’язків супроводжується розвитком псевдобульбарного синдрому, що підсилює явища дизартрії, що сприяє появі гримас насильницького плачу або сміху та інших симптомів. Порушення чутливості не властиві БАС. Контроль тазових органів зазвичай не порушується, що дозволяє диференціювати БАС від подібних поразок, викликаних сдавленней верхніх відділів спинного мозку.
Додаткові методи дослідження, за винятком міографії, що дозволяє виявити фасцікуляціі на ранній стадії або їх поширеність на більш пізніх стадіях захворювання, мало що дають для діагностики захворювання. Рівень білка в лікворі нормальний або злегка підвищений.
У деяких випадках відзначається підвищення рівня ферменту фосфокінази.
Захворювання носить прогресуючий характер, ремісії для нього не характерні. Залежно від переважної локалізації патологічного процесу розрізняють: шийно-грудний, попереково-крижовий, бульбарних форми БАС [Хондкаріан О. А., 1980]. Тривалість захворювання досягає 3-4 років, але іноді розтягується до 10 років і більше. Результат завжди фатальний. Хворі вмирають від виснаження, порушення дихання або приєдналася інфекції, найчастіше пневмонії.
Діагноз бічного аміотрофічного склерозу
Слід мати на увазі, що подібна до БАС картина може мати місце і при інших захворюваннях. Хронічно протікає остеохондроз шийних хребців в результаті здавлювання спінальних судин може викликати поступово наростаючу ішемію, в першу чергу в центральних відділах спинного мозку, передніх рогах і бічних стовпах, що супроводжується розвитком синдрому БАС. Рентгенографія шийного відділу хребта в таких випадках дозволяє виявити локальне ураження шийних хребців і звуження хребетного каналу. Схожа з БАС клінічна картина може бути обумовлена люетіческім процесом, але в даний час такі випадки зустрічаються вкрай рідко.
Деяка схожість з БАС можуть мати прогредієнтності форми кліщового енцефаліту. На користь останнього свідчать наявність гострого періоду, анамнестичні дані про укуси кліща, серологічні проби.
Лікування бокового аміотрофічного склерозу
Лікування носить симптоматичний характер. Показані помірний масаж, загальнозміцнюючі засоби. Короткочасне поліпшення може бути отримано при застосуванні вітаміну В12, прозерина, діазепаму (седуксену). При виникненні пневмонії показані антибіотики. Прогноз у всіх випадках бокового аміотрофічного склерозу несприятливий.
Захворювання вперше виділено в самостійну форму в 1901 р Г. І. Россолімо, а потім описано групою авторів в 1909 р і в 1912 р Синонімами цього захворювання є дистрофічна міотонія, або міотонічна дистрофія. Воно характеризується кількома синдромами: міотоніческім, Міопатичні, ендокринних і общедістрофіческім. Міотонічна синдром не відрізняється від такого при миотонии Томсена. Виразність його поступово зменшується в міру розвитку атрофії і парезів. У далеко зайшла стадії можна виявити лише характерну «ямку» в мові. Миопатический синдром характеризується поступовим розвитком атрофії м’язів з відповідним зниженням м’язової сили. Поширення атрофий при цьому своєрідне: завжди страждають м’язи обличчя, особливо верхньої його половини, жувальна мускулатура, м’язи шиї, особливо грудино-ключично-соскоподібного. В розгорнутій стадії процесу атрофії і парези поширюються на м’язи руки, особливо дистальнівідділи, і ноги, де грубо уражається перонеальная група м’язів з розвитком звисає стопи. Рано згасають сухожильні рефлекси.
Ендокринні і дистрофічні порушення проявляються зниженням активності гонад (імпотенція, ранній клімакс), наднирників (низький рівень 17-кетостероїдів, 17-оксикортикостероїдів), щитовидної залози, гіпофіза. Часто мають місце значне схуднення, раннє випадання волосся, зубів, витончення шкіри, специфічна точкова катаракта, низький артеріальний тиск, брадикардія. Як правило,
порушується провідність по передсердно-желудочковому пучку, спостерігаються аритмія, низький вольтаж на ЕКГ. При атрофической миотонии часто відзначається зміна інтелекту, іноді прояви олігофренії, своєрідне зміна характеру.
Перші ознаки хвороби виявляються частіше у віці 20-30 років, однак в сімейних випадках вдається їх виявити вже в 8-10 років. Іноді вже у новонароджених відзначається слабке смоктання, гіпотонія, уповільнене моторне і психічний розвиток. До певного віку стан може залишатися стабільним протягом ряду років, потім настає більш-менш швидке прогресування захворювання. У сім’ях хворих з розгорнутою симптоматикою дистрофічній миотонии у окремих членів можуть спостерігатися тільки поодинокі ознаки хвороби (міотонічна катаракта, ендокринні симптоми, зміна особистості та ін.).
и Атрофічна миотония — спадкове захворювання з аутосомно-домінантним типом передачі з високою пенетрантностью і варіабельнийекспресивністю.
Вроджена миотония описана вперше в 1874 р Leyden, а потім в 1876 р Thomsen, останній сам страждав цим захворюванням, так само як і багато членів його сім’ї.
Захворювання починається частіше всього у віці 10-12 років, в деяких сім’ях — з моменту народження або в більш пізньому віці. Протягом ряду років може спостерігатися поступове наростання симптомів, потім стан стабілізується. Захворювання відноситься до спадкових, передається по аутосомно-домінантним типом, частково обмеженого підлогою — чоловіки страждають у багато разів частіше. Becker вказує на можливість аутосомно-рецесивного варіанту захворювання, при якому спостерігається більш ранній початок і важча картина хвороби. Клінічна картина представлена міотоніческім феноменом майже в чистому вигляді: м’язове скорочення нормальне, проте розслаблення різко ускладнено, відбувається уповільнено, з великими труднощами через своєрідну м’язової контрактури або спазму. При повторенні однотипних рухів розслаблення стає все більш вільним, іноді навіть нормальним, проте після відпочинку все явища повторюються. Це складне становище рухів найбільш яскраво виражено в кистях і пальцях, в жувальної мускулатури, у важких випадках воно спостерігається в усіх м’язах. Слабкі і пасивні руху не викликають миотонической контрактури. Цей феномен носить назву «довільній миотонии». Характерно підвищення механічної збудливості м’язів: при ударі перкусійні молоточком в м’язі виникає «ямка» або «валик», які тримаються протягом досить тривалого часу. Особливо характерна «ямка» в мові, яка є патогномонічним симптомом. Характерний також симптом великого пальця — при ударі по піднесенню великого пальця останній наводиться і в такому положенні залишається деякий час. Подібний феномен носить назву «механічна миотония». При подразненні м’яза електричним струмом виникає «електрична миотония» або електрична міотонічна реакція з повільним розслабленням м’язи після виключення струму.
М’язова система у хворих зазвичай добре розвинена, хворі мають атлетичну статуру, однак м’язова сила щодо знижена. Сухожильнірефлекси нормальні або можуть бути відсутні, але при повторному дослідженні вони нормалізуються.
Важкість захворювання характеризується поширеністю міотоніческім порушень на різні м’язові групи, тривалістю фази розслаблення і часом повної ліквідації Міотонічна спазму при розробленні. У важких випадках може виникати генералізований миотонический спазм, що охоплює всю мускулатуру при спробі утримати рівновагу при раптовому поштовху або стрибку.
Хворі в цих випадках падають як підкошені і залишаються лежати нерухомо деякий час ( «ataxia muscularis», по Томсену).
Погіршення міотоніческім проявів відзначається на холоді, при фізичній перевтомі, поліпшення — в теплі . У жінок в період вагітності спостерігається різке погіршення, в період лактації симптоми майже зникають, а після її припинення повертаються до початкового рівня.
Ботулиническая полінейропатія характеризується також ураженням рухових функцій, однак місцем токсичного впливу не є нервові стовбури, а зони їх закінчень. Специфіка цього виду полінейропатії полягає в тому, що вона являє собою не ускладнення захворювання, а його обов’язкове прояв.
Ботулинический отрута — дуже сильний, завжди смертельний при зіткненні з мозком. Однак при отруєнні людини через рот отрута не проникає в ЦНС, так як він не долає гематоенцефалічний бар’єр. У спинномозковій рідині його не виявляють. Місцем дії ботулінічного отрути залишається лише зона нервово-м’язової передачі. У зв’язку з цим порушується механізм вивільнення ацетилхоліну, зменшується число його квантів (на відміну від міастенії, при якій зменшується кількість медіатора в кожному Кванті). Звідси — переважне ураження певних м’язів, головним чином окорухових, глоткових (по особливому реагують в умовах денервації на ацетилхолін), відсутність атрофії, збереження свідомості при наявності грубих порушень артикуляції, ковтання. Слід знову підкреслити, що в основі описуваного бульварного синдрому лежить медіаторний нервово-м’язовий дефект. Клінічні прояви починаються вже через кілька годин після вживання недоброякісних консервів, ковбас, в яких розмножується бацила ботулізму. З’являються блювання, пронос, болі в животі, сухість слизових оболонок і шкіри. Незабаром приєднуються параліч акомодації, рефлекторна нерухомість розширених зіниць, двоїння, розлади фонації, ковтання, слабкість мускулатури, перш за все шийної, при відсутності адекватного лікування в 50-60% випадків настає смерть. Починається регрес симптомів йде до повного одужання.
До геморагічного інсульту відносять крововиливу в речовина мозку (крововилив в мозок або паренхиматозное крововилив) і в подоболочечние простору (субарахноїдальні, субдуральна, епідуральні). Спостерігаються і поєднані форми крововиливу — субарахноїдальний-паренхіматозні, паренхиматозно-субарахноїдальні і паренхиматозно-вентрикулярні.
Крововилив в мозок
Етіологія крововиливів в мозок
Крововилив в мозок найбільш часто розвивається при гіпертонічній хворобі, а також при артеріальній гіпертензії, обумовленої захворюваннями нирок, залоз внутрішньої секреції (феохромоцитома, аденома гіпофіза) і при системних судинних захворюваннях алергічної та інфекційно-алергічної природи, що супроводжуються підвищенням артеріального тиску (вузликовий періартеріїт, червоний вовчак). Крововилив в мозок може виникнути при вродженої ангіомі, при мікроаневрізми, що сформувалися після черепно-мозкової травми або септичних станів, а також при захворюваннях, що супроводжуються геморагічним діатезом — хвороби Верльгофа, лейкоз і уремії.
Патогенез крововиливів в мозок
В даний час визнано, що в патогенезі геморагій найбільше значення має артеріальна гіпертензія. Гіпертонія, особливо при гіпертонічній хворобі, веде до зміни судин, фібриноїдної дегенерації і гіалінозу артерій нирок, серця, а також всередині церебральних артерій. Зміни судин проходять кілька стадій: субендотеліальними серозна інфільтрація з підвищеною проникністю ендотелію для плазми крові супроводжується периваскулярной транссудацией і сприяє в подальшому концентрическому ущільнення стінок судин за рахунок фібриноїдної субстанції. Швидкий розвиток фібриноїдної дегенерації веде до формування розширених артеріол і до аневризмі. При цьому можна спостерігати, що елементи крові проникають в надірвані структури артеріальних стінок, і в цих місцях можуть утворюватися тромбози. В результаті Фібриноїдне-гиалиновой дегенерації артеріальних стінок можуть розвиватися розшаровуючі аневризми, які і вважають причиною кровотечі (per rexis) в результаті розриву судини.
[Рис. 1] Геморагічний інсульт, схематичне зображення
Інтенсивність і розміри мозкового кровотоку визначаються розміром аневризми, тиском випливає з неї крові і швидкістю її тромбування. Найбільш часто крововилив розвивається в області підкіркових вузлів, в області putamen з стріарних артерій.
Кровоизлияния в переважній більшості випадків розвиваються у хворих на гіпертонічну хворобу і при всіх інших захворюваннях, які супроводжуються артеріальною гіпертонією. При атеросклерозі без артеріальної гіпертензії крововиливи зустрічаються дуже рідко. При захворюваннях, що не супроводжуються артеріальною гіпертонією (хвороби крові, соматичні захворювання, що супроводжуються геморагічним діатезом, уремія та ін.), Основним механізмом розвитку геморагії є диапедез внаслідок підвищення проникності стінок судин.
Патологічна анатомія крововиливів в мозок
Крововилив в мозок розвивається частіше в результаті розриву судини і значно рідше внаслідок діапедезу.
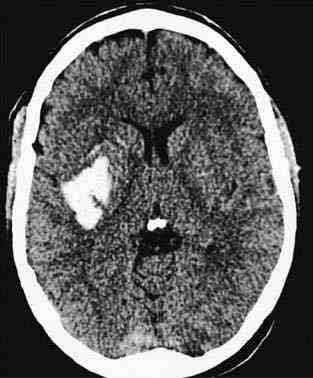
Морфологічно розрізняють гематоми, т . е. порожнини, заповнені рідкою кров'ю і згустками, добре відмежовані від навколишньої тканини, і геморагії з нерівними контурами, чітко не відокремленого — геморрагическое пропотеваніе. Звертає увагу переважна локалізація гематом в області підкіркових вузлів півкуль мозку. Значно рідше гематоми розвиваються в області зубчастих ядер мозочка і ще рідше -у області моста мозку. Формування гематоми відбувається головним чином внаслідок розсовування вилилась кров'ю мозкової речовини і здавлення останнього.
При крововиливі в мозок в 85-90% випадків спостерігається прорив крові в шлуночкову систему або в субарахноїдальний простір. Найбільш типове місце прориву — латерально-базальна частина переднього рогу бічного шлуночка (головка хвостатого ядра). Зустрічаються крововиливи з одномоментним проривом, стінки в різних ділянках шлуночкової системи.
При крововиливах по типу гематоми нерідко виявляють великий набряк мозку, сплощення звивин і розвиток грижових вклиненням мозку. Гематома полушарной локалізації викликає зміщення стовбура мозку з вклиненням його в тенторіальное отвір, наслідком чого є деформація стовбура мозку і розвиток в ньому вторинних дрібних крововиливів.
Кровоизлияния типу геморагічного просочування виникають переважно в зорових горбах, рідше в мосту мозку і білій речовині великих півкуль. Вони є результатом злиття дрібних вогнищ крововиливів, що виникають шляхом діапедезу з дрібних судин.
Класифікація крововиливів в мозок
У клінічній практиці широке поширення набула класифікація крововиливів в залежності від локалізації вогнища крововиливу. Серед паренхіматозних крововиливів розрізняють крововиливи у великі півкулі головного мозку, крововиливу в стовбур мозку і в мозочок. За локалізацією в півкулях крововиливи діляться на латеральні — назовні від внутрішньої капсули, медіальні — досередини від неї і змішані, що займають всю область підкіркових гангліїв.
Клініка крововиливів в мозок
Крововилив розвивається, як правило, раптово, зазвичай днем, в період активної діяльності хворого, хоча в поодиноких випадках спостерігаються крововиливи як в період спокою хворого, так: і в час сну. Для геморагії в головний мозок характерне поєднання загальномозкових і вогнищевих симптомів.
Раптова головний біль, блювота, порушення свідомості, прискорене гучне дихання, тахікардія з одночасним розвитком гемиплегии або геміпарезу — звичайні початкові симптоми крововиливу. Ступінь порушення свідомості буває різною — від легкого оглушення до глибокої атонической коми. При визначенні глибини розлади свідомості звертають увагу на можливість контакту з хворим, виконання хворим простих і складних вказівок, можливість повідомити анамнестичні відомості, швидкість і повноту відповідей хворого, збереження критики, ставлення до свого стану, орієнтуванні хворого в навколишньому середовищі. При глибокої непритомності мовного контакту з хворим немає, лише фіксується реакція хворого на гучні звуки, на укол або серію уколів.
При легкому ступені оглушення як у відповідях на питання, так і при виконанні наказів (навіть якщо у хворого немає афазії), видно сповільненість реакцій, збільшення латентного періоду. Виконує складні вказівок хворому не вдається, він швидко «виснажується.» І «відключається», хоча може повідомити про себе відомості, але плутає їх, відповідає на питання уповільнено і «невпопад». Нерідко відзначаються рухове занепокоєння, тривога, недооцінка свого стану, реакція на укол збережена — наголошується гримаса болю і вилучання руки або ноги.
відзначають в початковому періоді оглушення або сопор можуть через кілька годин перейти ВКОМ . Кома характеризується більш глибоким порушенням всіх життєво важливих функцій (дихання, серцевої діяльності), зниженням або втратою реакцій на подразники. Хворий не реагує на одиночний укол, слабкі і середні звуки, на дотик, але відсмикує здорову руку у відповідь на серію уколів. При атонической комі — крайнього ступеня термінального стану — втрачаються всі рефлекси (зрачковие, корнеальні, глотковий, шкірні, сухожилкові), артеріальний тиск падає, змінений ритм дихання — дихання типу Чейна -I Стокса змінюється диханням типу Куссмауля. Характерний загальний вигляд хворого з масивним крововиливом в півкулі: очі закриті, шкірні покриви гіперемійовані, нерідко спостерігається рясне потовиділення. Пульс напружений, артеріальний тиск підвищений. Очі повернені в сторону ураженого півкулі (параліч коркового центру погляду), зіниці можуть бути різної величини (анізокорія зустрічається в 60-70% випадків крововиливів полушарной локалізації), зазвичай зіницю більше на стороні вогнища. Нерідко відзначається розходиться (косоокість, обумовлене, як і анізокорія, здавленням окоруховогонерва на боці гематоми, яке є симптомом, що вказує на розвивається здавлення стовбура мозку гематомою і перифокальним набряком мозку, спочатку виникли в півкулі, де стався крововилив.
Найбільш частий вогнищевий симптом крововиливи — геміплегія. Зазвичай поєднується з центральним парезом лицьової мускулатури і мови, а також темігіпестезіей в контралатеральних кінцівках і гемианопсией. До вогнищевих симптомів (Крововилив в великі півкулі головного мозку слід віднести параліч погляду, сенеорно-моторну афазію (при левополушарной локалізації крововиливу), анозогнозію, т. Е. Неусвідомлення хворим свого паралічу, при крововиливі в праву півкулю. При крововиливах в праву півкулю спостерігаються насильницькі руху в здорових правих кінцівках — паракінези або автоматизовані руху. Паракінези можна відзначити незабаром після інсульту в фазі психомоторного збудження, коли свідомість втрачено ще не повністю. Хворий рухає здоровою рукою і ногою, як би жестикулюючи, або доторкається до носа, підборіддя, чухає живіт, згинає і розгинає ногу. Зовні ці рухи нагадують цілеспрямовані, проте у міру порушення свідомості вони стають все більш автоматизованими.
Значне місце в клініці гострого періоду крововиливи займають дистонії, різні варіанти порушення м'язового тонусу, детально вивчені вітчизняними невропатологами: С. Н. Давиденкова (1921), Н. К. Боголєпова (1953), Д. К. Луньовим (1962) та ін. У перший момент гостре порушення мозкового кровообігу веде до розвитку м'язової гіпотонії на стороні паралічу.
Підвищення м'язового тонусу може розвинутися безпосередньо після інсульту або через кілька годин або навіть кілька днів. Для крововиливу в мозок найбільш характерно підвищення м'язового тонусу приступообразно, у вигляді пароксизмів. Нападоподібне підвищення м'язового тонусу, назване С. Н. Давиденкова горметонія, клінічно проявляється дуже яскраво.
Пароксизмальне підвищення м'язового тонусу частіше спостерігається в паралізованих кінцівках, однак воно може бути і в гомолатеральних вогнища кінцівках. В руках тонічний спазм зазвичай охоплює аддуктори плеча, флексоров і пронатори передпліччя, в ногах — аддуктори стегна, екстензоров гомілки і внутрішні ротатори стопи. Можна спостерігати, як у міру розслаблення тонічних спазмів у зазначених м'язах виникає підвищення м'язового тонусу в м'язах — антагоністах. Тривалість таких нападів м'язової гіпертонії коливається від декількох секунд до декількох хвилин. Напади горметонічеекіх судом посилюються від різних екстсро- і інтерорецептівних подразнень. Іноді судоми горметонии досягають такої інтенсивності, що супроводжуються переміщенням кінцівки. У деяких хворих спостерігається часткова горметония, т. Е. Що охоплює будь-яку одну кінцівку, у інших — гемігорметонія.
Особливо різко пароксизмальное підвищення м'язового тонусу спостерігається при нолушарних крововиливах, що супроводжуються проривом крові в шлуночки мозку. Зміна м'язового тонусу при полушарних крововиливах пов'язано з дисфункцією тонігенних структур мозкового стовбура, що регулюють м'язовий тонус, обумовленої сдавленней і дислокацією стовбура.
При паренхіматозних крововиливах ж через кілька годин (іноді до кінця першої доби) з'являються менінгеальні симптоми. При цьому ригідності потиличних м'язів може не бути зовсім, рідко викликається верхній симптом Брудзинського, але з великою постійністю відзначаються симптом Керніга на непаралізованной стороні і позитивний нижній симптом Брудзинського. Відсутність симптому Керніга на стороні паралічу служить одним з критеріїв визначення боку ураження.
Підвищення температури тіла спостерігається у хворих з паренхіматозним крововиливом через кілька годин з моменту захворювання і тримається кілька днів в межах 37-38 ° С. При прориві крові в шлуночки і при близькості вогнища крововиливу до гіпоталамічної області температура тіла досягає 40-41 ° С. Як правило, в периферичної крові спостерігається лейкоцитоз, невеликий зсув лейкоцитарної формули вліво, в першу добу захворювання відзначається підвищений вміст цукру, іноді залишкового азоту. Нерідко відзначається підвищена фібринолітична активність крові, в більшості випадків знижена агрегаціятромбоцитів.
Перебіг і прогноз крововиливів у мозок
При церебральних геморрагиях відзначається велика летальність, яка за даними різних авторів коливається між 75-95%. До 42-45% хворих з масивним крововиливом в головний мозок помирають протягом 24 годин від початку інсульту, інші вмирають на 5-8-у добу захворювання і в рідкісних випадках на 15-20-ту добу. Найбільш частою причиною смерті хворих з геморагічними інсультами є обмеження стовбура при полушарной геморагії в зв'язку з набряком мозку. Друге місце по частоті причин смерті займає саме вогнище з масивним проривом крові в шлуночкову систему і руйнуванням життєво важливих утворень.
Лікування крововиливів в мозок
Хворого з крововиливом в мозок необхідно правильно укласти в ліжко, надавши голові піднесене положення, піднявши головний кінець ліжка. При крововиливі в мозок перш за все необхідна терапія, спрямована на нормалізацію вітальних функцій, зупинку кровотечі і на боротьбу з набряком мозку, а потім вирішення питання про можливість видалення крові, що вилилася.
Перш за все слід забезпечити вільну прохідність дихальних шляхів, для чого необхідно видалити рідкий секрет з верхніх дихальних шляхів за допомогою спеціальних відсмоктувачів, застосувати ротові і носові повітроводи, протерти ротову порожнину хворого. При супутньому набряку легенів рекомендуються кардиотоники: 1 мл 0,06% розчину Карг чи, кін а чи 0,5 мл 0,05% розчину строфантину з глюкозою в / в, а також вдихання кисню з парами спирту з метою зменшення ціноутворення в альвеолах. Призначають атропін 1 — 0,5 мл 0,1% розчину, фуросемід (лазикс) 1-2 мл 1% розчину, димедрол 1 мл 1% розчину в / м.
Необхідно застосування коштів, спрямованих на попередження та усунення гіпертермії. При температурі тіла близько 39 ° С і вище призначають 10 мл 4% розчину амідопірину або 2-3 мл 50% розчину анальгіну в / м. Рекомендується також регіонарна гіпотермія великих судин (бульбашки з льодом на область сонних артерій на шиї, в пахвові і пахові області).
Щоб зупинити кровотечу і попередити його відновлення, необхідно знизити артеріальний тиск і підвищити згортання крові. Для зниження артеріального тиску використовують дибазол (2-4 мл 1% розчину), гемитон (1 мл 0,01% розчину). При відсутності ефекту призначають аміназин (2 мл 2,5% розчину і 5 мл 0,5% розчину новокаїну) в / м або в складі суміші: аміназин (2 мл 2,5% розчину), димедрол (2 мл 1% розчину) , промедол (2 мл 2% розчину) в / м, гангліоблокатори — пентамін (1 мл 5% розчину в / м або 0,5 мл в 20 мл 40%) розчину глюкози в / в, повільно під контролем АТ), бензогексоній ( 1 мл 2% розчину в / м), арфонад (5мл 5% розчину в 150 мл 5% розчину глюкози в / в зі швидкістю 50-30 крапель в хвилину). Гіпотензивні препарати повинні застосовуватися з обережністю.
Гангліоблокатори можуть різко знизити артеріальний тиск, тому призначати їх слід у виняткових випадках, при АД, що перевищує 200 мм рт. ст. Вводити гангліоблокатори треба обережно з постійним наглядом за АТ кожні 20-30 хв. При цьому слід домагатися зниження тиску до оптимального рівня, індивідуального для кожного хворого.
Показані засоби, що підвищують згортання крові і зменшують судинну проникність: 2 мл 1% розчину вікасолу, препарати кальцію (10 мл 10% раствоpa хлориду кальцію в / в або глюконат кальцію 10 мл 0,25% розчину в / м). Застосовують 5% розчин аскорбінової кислоти — 5-10 мл в / м.
Хворим з геморагічним інсультом необхідно призначати препарати, які гальмують патологічно підвищену фібринолітичну активність крові. З цією метою застосовують амінокапронову кислоту, вводячи ер у вигляді 5% розчину в / в крапельно по 100 мл під контролем вмісту фібриногену і фібринолітичної активності крові протягом перших двох діб. Для зниження внутрішньочерепної гіпертензії і зняття набряку мозку застосовують фуросемід — лааікс (20-40 мг в / в або в / м), а також мавніт (готовий 10-15-20% розчин з розрахунку 1 г в 200 мл ізотонічного розчину хлориду натрію або 5% розчину глюкози в / в крапельно). Небажано застосування сечовини, так як наступає слідом за потужним протинабрякову ефектом викарное розширення судин мозку може привести до повторного ще більш грубому набряку і можливого кровотечі в паренхіму мозку. Дегідратаційним дію має гліцерин, що підвищує осмотичний тиск крові, що не викликає порушення електролітного балансу.
Інфузійну терапію слід проводити під контролем показників кислотно-лужної рівноваги та електролітного складу плазми. При наростанні набряку мозку і загрозу життю хворого показано хірургічне лікування.
Хірургічне лікування. Оперативне втручання при внутрішньомозкової гематоми зводиться до видалення крові, що вилила і створення декомпресії. В даний час накопичений багаторічний досвід хірургічного лікування геморагічних інсультів. Можна вважати загальновизнаною точку зору нейрохірургів, що хірургічне лікування показано при латеральних гематомах і недоцільно при медіальних і великих крововиливах. Хірургічне лікування при латеральних гематомах доцільно проводити в першу добу інсульту до розвитку зсуву, деформації та здавлення стовбура мозку. При хірургічному лікуванні гематоми летальність у порівнянні з консервативною терапією знижується з 80% до 50-40% [Арутюнов А. І., Ромоданов А. П., Педаченко Г. А., 1967, Богатирьов Ю. В., 1968].
Субарахноїдальнийкрововилив
Етіологія субарахноїдальних крововиливів
В більшості випадків причиною спонтанного субарахноїдального крововиливу є розрив внутрішньочерепної аневризми. Артеріальні аневризми головного мозку, як і аневризми іншої локалізації, являють собою обмежене або дифузне розширення просвіту артерії або випинання її стінки. Більшість аневризм артерій головного мозку має характерний вигляд невеликого тонкостінного мішка, в якому зазвичай можна розрізнити дно, середню частину і так звану шийку.
У зв'язку з цими анатомічними особливостями подібні аневризми часто називають мішкоподібними. Рідше аневризма має вигляд великого сферичного освіти або ж дифузного розширення артерії на значному протязі (так звані S-образні аневризми). Більшість аневризм розташовується на артеріях основи мозку. Їх улюбленою локалізацією є місця розподілу і анастомозірованія артерій мозку. Особливо часто аневризми локалізуються на передній сполучної артерії в місці відходження задньої сполучної артерії від внутрішньої сонної артерії або в області гілок середньої мозкової артерії. Порівняно невелика частина аневризм локалізується в системі вертебральних і базилярної артерій. У жінок аневризми бувають частіше, ніж у чоловіків.
Питання про походження мішечкуваті аневризм, які складають переважну більшість аневризм, до теперішнього часу залишається в значній мірі відкритим. На думку більшості авторів, в основі освіти аневризм лежать дефекти розвитку судинної системи мозку, інша (менш численна) група дослідників підкреслює роль атеросклерозу і гіпертонічної хвороби як однієї з головних причин виникнення мішечкуваті аневризм.
концепція травматичного генезу аневризм судин головного мозку запропонована М. Б. Копиловим (1962), який вважає, що в момент травми різко зростає тиск в артеріях мозку. Під впливом такого гемогідравлічеокого удару може виникнути пошкодження артеріальної стінки з подальшим розвитком аневризми. Невелика частина аневризм розвивається в зв'язку з попаданням в артерію мозку інфікованих емболів. Ці так звані мікотіческіе аневризми характеризуються переважним розташуванням на конвекситальной поверхні мозку. Вони найчастіше розвиваються у людей молодого віку, які страждають затяжним септичним ендокардитом. У походження великих сферичних і S-образних аневризм безсумнівно провідну роль відіграє атеросклероз.
Далеко не всі аневризми викликають клінічні симптоми. Велика частина аневризм є випадковою знахідкою при патологоанатомічному дослідженні. Аневризми виявляють у людей різного віку — від дитячого до старечого. Клінічно аневризми проявляють себе субарахноїдальним крововиливом на четвертому і п'ятому десятилітті життя.
Серед інших причин субарахноїдального крововиливу відзначають атеросклеротичні і гіпертонічні зміни судин, пухлини мозку первинні і метастатичні, запальні захворювання, уремию, хвороби крові.
Клініка субарахноїдальних крововиливів
Зазвичай субарахноїдальний крововилив розвивається раптово, без будь-яких провісників. Лише у невеликої частини хворих до крововиливу спостерігаються симптоми, обумовлені наявністю аневризми — обмежені болю в лобно-очноямкову області, парези черепних нервів (частіше окоруховогонерва). Розрив аневризми може статися в момент фізичного або емоційного напруження.
Перший симптом субарахноїдального крововиливу — раптовий гострий головний біль, яку самі хворі визначають як «удар», як відчуття «поширення в голові гарячої рідини». У перший момент захворювання біль може мати локальний характер (в області чола, потилиці), потім стає розлитої. Надалі у хворого з'являється біль в шиї, в спині і ногах. Майже одночасно з головним болем виникають нудота, багато кратна блювота. Слідом за нападом головного болю настає втрата свідомості. У легких випадках втрата свідомості нетривала (10-20 хв), у важких — несвідомий стан триває багато годин і навіть діб. У момент розриву аневризми або незабаром після нього можуть спостерігатися епілептичні припадки.
Для крововиливів з артеріальних аневризм особливо характерно швидкий розвиток менінгеального симптомокомплексу. При обстеженні хворого виявляються ригідність потиличних м'язів, симптоми Керніга і Брудзинського, світлобоязнь, загальна гіперестезія. Лише у найбільш важко хворих з пригніченням рефлекторної діяльності менінгеальні симптоми можуть бути відсутні.
Частим симптомом, супроводжуючим субарахноїдальний крововилив, є порушення психіки. Ступінь психічного розладу може бути різною-від невеликої сплутаності, дезорієнтацію до важких психозів. Нерідко після крововиливу спостерігається психомоторне збудження або розвиваються порушення пам'яті, характерні для корсаковского синдрому.
Як реакція на излившуюся кров в подоболочечное простір, а також в результаті роздратування гіпоталамічної області, в гострому періоді спостерігається підвищення температури тіла до 38-39 ° С, зміни з боку крові у вигляді помірного лейкоцитозу і зсуву лейкоцитарної формули вліво. Поряд з цим у багатьох хворих, які не страждають на гіпертонічну хворобу, спостерігається підйом артеріального тиску. У важких випадках, при масивних крововиливах, спостерігаються виражені порушення вітальних функцій: серцево-судинної діяльності та дихання.
У гострій стадії субарахноїдального крововиливу ряд симптомів обумовлений швидким підвищенням внутрішньочерепного тиску (головний біль, блювота). Підвищення внутрішньочерепного тиску і викликане цим утруднення венозного відтоку призводять до розвитку застійних явищ на очному дні. Крім розширення вен і набряклості сосків зорових нервів можуть бути виявлені крововиливи в сітківку ока.
У великому відсотку випадків при субарахноїдальний крововилив спостерігаються також парези черепних нервів і симптоми вогнищевого ураження мозку. Поразки черепних нервів у хворих зі спонтанними субарахноїдальними крововиливами є патогномонічними для розриву базальних артеріальних аневризм. Найчастіше спостерігається ізольований парез окорухового нерва, що виникає в момент розриву аневризми або незабаром після нього. У переважній більшості випадків ізольоване однобічне ураження окорухового нерва спостерігається при крововиливі з аневризми, що розташовується в місці відходження задньої сполучної артерії від внутрішньої сонної. Кровоизлияния з аневризм внутрішньої сонної і передньої сполучної артерій поблизу зорових нервів і хіазми порівняно часто супроводжуються ураженням зору. Порушення функцій інших черепних нервів спостерігається рідше.
Відзначено дві основні причини поразки черепних нервів у хворих з артеріальними аневризмами. По-перше, безпосереднє здавлення нерва аневризмою і, по-друге, крововилив в нерв і його оболонки в момент розриву аневризми з подальшим утворенням сполучнотканинних периневральних зрощень.
У багатьох хворих розвиваються симптоми вогнищевого ураження мозку: парези кінцівок, порушення чутливості, мовні розлади тощо. Виникнення цих симптомів обумовлено найчастіше супутнім крововиливом в мозок або локальною ішемією мозку, викликаної спазмом артерій або ішемією, внаслідок тромбування аневризми.
Вивченню клінічних проявів артеріального спазму при розриві артеріальних аневризм, патологоанатомічним змінам в мозку, обумовленим спазмом, присвячене в даний час багато робіт. Судячи з ангіографічним даними, найбільш виражений спазм артерій відзначається поблизу аневризми, проте в окремих випадках можна виявити і спазм артерій, розташованих на віддалі від неї. Тривалість спастичного скорочення артерій найчастіше не перевищує 2-4 тижнів.
Висловлюються припущення, що розвивається в результаті спазму гостра ішемія стовбура мозку є найбільш імовірною причиною ряду важких симптомів, які супроводжують розрив аневризми, таких , як втрата свідомості, порушення дихання та серцевої діяльності. Цікавим є те обставина, що артеріальний спазм може бути причиною не тільки ішемії мозку поблизу розірвалася аневризми, а й віддаленого полушарного поразки. Так, при аневризмах передньої сполучної артерії часто вдається виявити локальні симптоми, викликані порушенням кровообігу в басейні передніх мозкових артерій — парези ніг, психічні зміни, дефекти праксису. Спазм середньої мозкової артерії призводить до парези протилежних кінцівок, порушення чутливості в них і афатические явищам. Причини спазму артерій при розривах артеріальних аневризм вивчені недостатньо. Висловлюється припущення, що велике значення мають такі фактори, як пошкодження стінки артерії і її сегментарного нервового апарату токсичними продуктами розпаду формених елементів крові.
Перебіг і прогноз субарахноїдальних крововиливів
Прогноз внутрішньочерепних крововиливів, викликаних розривом артеріальних аневризм, несприятливий. У більшості випадків справа не обмежується одноразовим крововиливом. Повторні крововиливи з аневризм протікають особливо важко. При них частіше спостерігаються парези, паралічі, а смертність приблизно вдвічі більше, ніж при первинних крововиливах.
Спостереження за хворими, які перенесли субарахноїдальний крововилив, дозволило встановити, що основна частина рецидивів припадає на 2 4-й тиждень після першого крововиливу. Через 2 місяці після розриву аневризми повторні крововиливи виникають порівняно рідко. Протягом перших 4-6 тижнів помирає до 60% хворих з крововиливами з артеріальних аневризм.
Діагноз субарахноїдальних крововиливів
Клініка субарахноїдального крововиливу можна вважати добре вивчена, і в типових випадках діагноз не викликає серйозних труднощів. Однак в ряді випадків на початку захворювання, коли менінгеальний симптомокомплекс ще не повністю розвинувся і на перший план виступають такі симптоми, як блювота, головний біль, підвищення температури, помилково може бути поставлений діагноз гострої інтоксикації, харчового отруєння. В інших випадках при відносно м'якому поступовому розвитку синдрому субарахноїдального крововиливу виникає підозра на цереброспінальної менінгіт. Діагностичні труднощі в більшості випадків можуть бути легко усунені за допомогою пункції. Діагноз субарахноїдального крововиливу вважається підтвердженим при наявності крові в спинномозковій рідині. Дослідити рідину потрібно швидко, щоб уникнути (неправильних терапевтичних заходів.
У перші дні після субарахноїдального крововиливу спинномозкова рідина більш-менш інтенсивно забарвлена кров'ю. Однак макроскопічний аналіз кривавої рідини не є достатнім для підтвердження діагнозу. Взяту рідина рекомендується центрифугувати. В отриманій після центрифугування рідини при субарахноїдальний крововилив визначається ксантохромия. Крім того, діагноз субарахноїдального крововиливу в перші години захворювання можна підтвердити наявністю вилужених еритроцитів при мікроскопічному дослідженні спинномозкової рідини. Через добу і більше після субарахноїдального крововиливу в ній з'являються макрофаги і лімфоцитарний цитоз.
Остаточний діагноз аневризми артерій головного мозку, визначення її точної локалізації, форми і розміру можливі тільки за допомогою ангіографії. Навіть найретельніше неврологічне обстеження в більшості випадків дозволяє лише з більшою або меншою ймовірністю припускати аневризму артерій мозку, а точна топічна діагностика аневризм, особливо множинних, практично неможлива. Небезпеки, пов'язані з транспортуванням хворого в спеціалізовані нейрохірургічні стаціонари, у багатьох випадках перебільшені. Часто труднощі для диференціального діагнозу виникають при субарахноїдальних крововиливах з пухлин мозку, до цього що протікають безсимптомно. Однак наростання вогнищевих симптомів ураження речовини мозку, а також нейтрофільний цитоз в спинномозковій рідині, що спостерігається протягом всього періоду захворювання при пухлинах мозку, дозволяють поставити правильний діагноз.
Лікування субарахноїдальних крововиливів
Лікування субарахноїдальних крововиливів включає консервативні і хірургічні методи в залежності від етіології, що викликала подоболочечное крововилив.
Необхідно дотримання суворого постільного режиму протягом 6 тижнів. Тривалість цього терміну пов'язана з тим, що переважна більшість повторних крововиливів з аневризм буває протягом 1-11 / 2 міс після першого. Крім того, значний період потрібно для утворення міцних сполучнотканинних зрощень поблизу розірвалася аневризми.
У гострій стадії крововиливу з метою створення умов для тромбування аневризми показані препарати, що підвищують згортання крові (вікасол, хлорид кальцію) , а також кошти, спрямовані на пригнічення фібринолітичної активності крові. З цією метою застосовують амінокапронову кислоту 10-15 г щодня протягом перших 3-6 тижнів, небезпечних для повторного крововиливу.
Оскільки навіть незначне напруження або хвилювання може викликати підвищення артеріального тиску і спровокувати повторний крововилив, необхідно застосування седативних засобів. Призначення цих препаратів в гострому періоді крововиливу тим більше показано, що багато хворих, які перенесли крововилив з аневризм, бувають порушені. При сильному збудженні потрібне застосування таких препаратів як діазепам (седуксен), аміназин та ін. Важливо контролювати роботу кишечника.
Розрив аневризми часто супроводжується підвищенням артеріального тиску, тому виникає необхідність в призначенні засобів, що знижують рівень артеріального тиску. У тих випадках, коли розрив аневризми супроводжується поширеним і стійким спазмом артерій мозку, виникає необхідність в застосуванні препаратів, що знімають спастичне скорочення артерій і поліпшують колатеральний кровообіг. На жаль, існуючі судинорозширювальні засоби малоефективні при артеріальному спазмі, спричиненому розривом аневризми.
Показана також терапія, спрямована на боротьбу з набряком мозку і внутрішньочерепної гіпертензією. З цією метою застосовують повторні спинномозкові пункції, салуретики, гліцерин і барбітурати парентерально.
Радикальним методом лікування артеріальних аневризм є хірургічний. Оперативне втручання до недавнього часу вважали показанням для попередження повторного крововиливу, що розвивається, як правило, протягом 2-6-го тижня після розриву аневризми. Однак в останні роки це питання переглядається, так як за даними ряду спостережень консервативна терапія, спрямована на пригнічення фібринолізу і проводиться протягом терміну, небезпечного для розвитку рецидиву, надійно попереджає повторні субарахноїдальні крововиливи.
«Ми те, що ми їмо» — говорить відома прислів'я, і вчені постійно доводять правоту цієї думки. Так, нещодавно співробітники Life Sciences Research Organization встановили, що ймовірність небезпечних для життя захворювань — інсульту та інфаркту — можна значно знизити, просто вживаючи в їжу лісові горіхи. Основою для даного наукового висновку ст
[2015.09.02]
Вчені з Нідерландів (а саме, співробітники університету Еразмуса) зробили дещо несподівані висновки, грунтуючись на проведеному дослідженні. Виявилося, що робота нирок має прямий зв'язок з роботою мозку, тому їх здорове функціонування важливо для розумових можливостей людини. Підставою для такого погляду стало
Внутрішньоклітинні ліпоїдози — група спадкових порушень обміну ліпідів, що супроводжуються їх підвищеною накопиченням в нервової тканини і ретикулоендотеліальної системі.
Патогенез і патологічна анатомія внутрішньоклітинних Ліпоїдоз
Спільною рисою цієї групи захворювань, що включають хвороба Гоше, хвороба Німана — Піка, ідіотії Тея — Сакса, є зміна сфинголипидов — ліпідів, що містять аміноспирт сфингозин [ Jatzkewitz, 1969]. Основні зміни при хворобі Гоше відбуваються в обміні глікоцереброзідов внаслідок інактивації ензиму глікоцереброзідази [Brady et al., 1965, 1966]. Глікоцереброзіди накопичуються в різних органах і тканинах, особливо в ретикулоендотеліальної системі, а при інфантильною формою і в нервових клітинах. При хвороби Гоше відзначені зміни в обміні білків, зокрема — аминоацидурия. У головному мозку у хворих Ліпоїдоз, як, наприклад, при інфантильною формою хвороби Гоше, виявляють відкладення ліпідів в гангліозних клітинах і клітинах глії, недостатність мієлінізації. В органах з підвищеним вмістом цереброзидів знаходять клітини Гоше. За хімічним складом цереброзидів при хвороби Гоше значно відрізняються від норми: до 70% вуглеводів становить глюкоза, в той час як в нормі переважає галактоза. Ідентичні особливості патогенезу встановлені при хворобі Німана — Піка. Основні зміни ліпідів стосуються сфінгоміеліна, поряд зі Сфінгомієлін в органах і тканинах хворих відкладається холестерин (з цих позицій хвороба Німана — Піка може вважатися сфингомиелин-холестеринових ліпоїдоз). Морфологічно при хворобі Німана — Піка виявляють збільшення маси і ущільнення речовини головного мозку, стушеванность кордону між сірим і білою речовиною. Спинний мозок також щільний, зі стертим малюнком. Виявлено грубі зміни в печінці, селезінці та інших органах. Клітини в корі головного мозку баллонообразное роздуті, містять велику кількість ліпідів, які є Сфінгомієлін, що підтверджується забарвленням по Шпільмейєра. Проліферація глії виражена помірно. У легенях, печінці, зобної залозі, селезінці виявлені клітини Піка — характерні ознаки захворювання. Клітини містять багато ліпідів і фарбуються відповідними фарбами. Первинним ензиматичними дефектом при хворобі Тея-Сакса є дефіцит гексозамінодази А, в результаті чого грубо порушується обмін ганглиозидов, в тканинах організму збільшується кількість гангліозиду GM2. При ювенільної формі ідіотії активність ферменту гексозамінодази А знижена в меншій мірі і накопичення гангліозиду GM2 також висловлено не настільки грубо. При вродженої формі ідіотії відзначається накопичення гангліозиду GM3. У головному мозку хворих амавротіческой ідіотією Тея-Сакса міститься велика кількість пінистих клітин, що містять різні ліпіди, в процес накопичення втягуються нейрони і глия. Вивчення нервових клітин при ювенільної формі ідіотії виявило складний хімічний склад накопичуються ліпідів і суттєва відмінність ультраструктури гранул в порівнянні з інфантильною формою ідіотії. Таким чином, хвороба Тея-Сакса представляє гангліозідний липоидоз, інші форми «amaurotics» — більш легкі варіанти порушення обміну ліпідів. Поразка клітинних елементів в головному мозку при ідіотії Тея — Сакса комбінується з помірно вираженими ознаками порушення мієлінізації. У білій речовині мозку значно зменшується вміст цереброзидів і сульфатідов — типових ліпідів мієліну. Біохімічні порушення не обмежуються нервовою системою, так як гангліозиди можуть міститися в печінці і селезінці хворих, накопичуючись в пінистих клітинах, які виявляються в цих органах. Макроскопічні зміни в нервовій системі у хворих з амавротіческой ідіотією Тея — Сакса виражені переважно в головному мозку — він збільшений в розмірах, шлуночки розширені, сіра речовина великих півкуль стоншена. Біла речовина щільне. Чітка картина склерозу головного мозку з атрофією сірої речовини, атрофія зорових нервів і трактів — найбільш типові риси морфологічних змін при цьому захворюванні. Гістологічно виявлені перицелюлярний набряк кори великих півкуль, скупчення клітин з ексцентрично розташованим ядром і вакуолінізірованной цитоплазмой переважно навколо судин, утворення клітин-куль у великій кількості, дистрофічні зміни нервових клітин виявлені в корі великих півкуль і підкіркових вузлах, стовбурі головного мозку, мозочку.
ідіотії Тея — Сакса
Клініка амавротіческой ідіопатія Тея — Сакса
Перебіг захворювання при всіх формах внутрішньоклітинних Ліпоїдоз відрізняється злоякісністю, поєднаним характером ураження нервової системи та інших органів і систем. Поряд із загальними ознаками, властивими всій групі внутрішньоклітинних Ліпоїдоз, виявлені симптоми, специфічні для окремих форм. Перші клінічні симптоми при ідіотії Тея — Сакса виявляються в кінці першого півріччя життя, коли діти перестають реагувати на навколишнє, грати, втрачають набуті навички. Виявляється феномен гиперакузии — різкі загальні рухові реакції у відповідь на слухове роздратування — поплескування в долоні, раптовий стукіт у двері. Виявляється падіння зору, дитина перестає фіксувати погляд на іграшках, блискучих предметах, в подальшому розвивається сліпота. У всіх хворих амавротіческой ідіотією Тея — Сакса, як правило, рано виявляються поліморфні судомні напади. Судоми частіше починаються з м'язів обличчя, посмикування очних яблук, в подальшому відзначаються тоніко-клонічні судоми кінцівок. Спостерігаються також зупинка погляду, салаамови судоми. Судоми зазвичай виражені досить симетрично в правій і лівій половині тіла, відрізняються стійкістю до проведеної протисудомну терапію. Особливістю судомних нападів в розгорнутій стадії хвороби є легкість їх виникнення під впливом зовнішніх подразників: дотики до дитини, різкого стуку. З перебігом хвороби напади стають частішими, збільшується їх тривалість. Хворі діти відрізняються підвищеною харчуванням, пастозністю. Виявляється гіперсалівація, сухість шкіри з гіпергідрозом стоп і долонь, гірсутизм.
Діагностика Тея — Сакса
Діагностика Тея — Сакса в розгорнутій стадії хвороби не становить труднощів. У початкових стадіях, коли у дітей спостерігаються судомні напади, зниження зору, підвищення температури тіла, часті блювоти, затримка психічного розвитку, нерідко ставляться діагнози: «енцефаліт», «наслідки родової травми», «спазмофилия». Для діагнозу має виняткове значення облік анамнестичних даних, послідовності розвитку симптомів, неухильного перебігу хвороби, даних додаткових методів дослідження, особливо офтальмологічних і біохімічних. Певну допомогу в діагностиці надає дослідження очного дна. В розгорнутій стадії хвороби в області макули відзначається сірувато-білий, злегка промінірующій круглий вогнище розміром в IV2-2 діаметра диска зорового нерва. Витончення сітківки носить дегенеративний характер і призводить до утворення яскраво-червоної плями в центрі макули, що обумовлено просвічує судинної оболонкою. Біохімічне дослідження ліпідів виявляє підвищення в плазмі крові рівня холестерину і цереброзидів і зниження в еритроцитах кількості сфінгоміеліна. Амавротическая ідіотія зустрічається 1: 250 ТОВ в популяції. Інші форми ідіотії зустрічаються значно рідше.
Вроджена форма Нормана-Вуда
Вроджена форма Нормана-Вуда клінічно проявляється в перші тижні життя дитини. Основними симптомами є: гідроцефалія (рідше-мікроцефалія), центральні парези та паралічі, судомні напади, косоокість, бульбарні розлади. Летальний результат наступає в найближчі місяці. Діагноз важкий, часом встановлюється тільки на розтині (накопичення гангліозиду GM3 і інших ліпідів в нервових клітинах).
Пізня дитяча форма Більшовского-янського
Пізня дитяча форма Більшовского-янського займає проміжне місце між інфантильною і ювенільної формами, внаслідок чого її нозологічна самостійність іноді ставиться під сумнів. Клінічні симптоми: судомні напади, атаксія, центральні парези, органічна деменція виникають в 3-4 роки, хвороба неухильно прогресує. На очному дні — атрофія зорових нервів. Летальний результат відзначається через кілька років від клінічного дебюту захворювання.
Юнацька форма Шпільмейєра — Фогта
Юнацька форма Шпільмейєра — Фогта частіше починається в кінці першого десятиліття життя. Характерно поєднання вираженої деменції і пігментного ретиніту. Виявляються парези кінцівок, псевдобульбарние симптоми, екстрапірамідні порушення. Часті судомні напади, нерідко переходять в епілептичний статус. Тривалість захворювання від декількох років до 10-15 років.
Пізня амавротическая ідіотія Куфса
Пізня амавротическая ідіотія Куфса виникає в зрілому віці і відрізняється повільним плином. Серед клінічних проявів провідними є прогресуюче зниження інтелекту, спастичний тетрапарез, псевдобульбарние симптоми, епілептичний синдром, пігментний ретиніт, атрофія зорових нервів. Хворі вмирають через 10-20 років від початку хвороби.
Ниманна-Піка хвороба
Клініка хвороби Німана — Піка
Перші симптоми інфантильною форми відзначаються у віці 5-6 мее. Діти стають млявими, малорухомими, у них зникає інтерес до навколишнього. Пізніше виявляються збільшення живота, схуднення, млявість змінюється періодами збудження. Відставання дитини у психофізичному розвитку посилюється. Іноді спостерігаються напади короткочасної асфіксії, загальні здригування, гіпертермічні кризи. Основними клінічними симптомами ураження нервової системи в розгорнутій стадії хвороби є відставання дітей в психічному і фізичному розвитку, помірна гідроцефалія, адинамія, що змінюються періодами занепокоєння, гіпертермічні кризи, парези кінцівок і напади асфіксії. Поряд з цим виявляються ознаки ураження внутрішніх органів — збільшення печінки та селезінки. Характерна ознака — часткова атрофія зорових нервів. Перебіг захворювання — неухильно прогресує. Подібна комбінація симптомів є підставою для діагнозу хвороби Німана — Піка, підтвердженням може служити виявлення на очному дні вишнево-червоного плямочки в макулярної області. На відміну від ідіотії Тея — Сакса вишнево-червона плямка в макулярної області виявляється приблизно в 1/3 випадків, від ідіотії хвороба Німана — Піка відрізняється також значною гепатоспленомегалией. Значно підвищується вміст сфінгоміеліна в спинномозковій рідині та крові хворих, що може бути використано в якості раннього діагностичного ознаки. Підвищується вміст холестерину в крові. Разом з тим є значні з-, трансформаційних змін інших фракцій фосфоліпідів, що обумовлено порушенням метаболізму ліпідів в клітинах різних органів і нервової системи.
Хвороба Гоше
Клініка хвороби Гоше
Клінічне вивчення хвороби Гоше дозволяє розмежовувати інфантильну і ювенильную її форми. Найбільш злоякісно протікає інфантильна форма хвороби Гоше: на першому році життя починає виявлятися затримка психічного і фізичного розвитку дитини. Звертають на себе увагу значне виснаження дитини і збільшення живота. Провідними симптомами є збільшення селезінки, в меншій мірі — печінки. Зміни з боку нервової системи проявляються затримкою нервово-психічного розвитку, підвищенням м'язового тонусу, косоокістю, порушенням терморегуляції. Дуже характерні зміни опорно-рухового апарату: кіфоз, деформація грудної клітки. Діти з інфантильною формою хвороби Гоше часто страждають пневмоніями. Летальний результат наступає в перші роки життя.
Ювенильная форма хвороби Гоше
Ювенильная форма хвороби Гоше виникає віці 2-4 років і відрізняється відносно м'яким перебігом. Домінують симптоми ураження внутрішніх органів (гепатоліенальнийсиндром і ін.), Анемія, геморагічні висипання на шкірі. Неврологічні прояви: слабо виражені пірамідні знаки, вегетативні симптоми (гіпергідроз, тахікардія, зміни дермографізму). Відзначається підвищення рівня цереброзидів в плазмі і еритроцитах. В кістковому мозку виявлені клітини Гоше. Клітини відрізняються фибриллярной структурою протоплазми, що дозволяє диференціювати їх від вакуолізірованние, пінистих клітин при хворобі Німана — Піка. Спадкування захворювання відбувається по аутосомно-домінантним типом на відміну від інфантильної форми хвороби Гоше, що передається по аутосомно-рецесивним типом.
Лікування хвороби Гоше
. Незважаючи на те що відомий первинний метаболічний дефект при окремих захворюваннях, відсутня ефективна терапія, хоча обговорюються спроби замісної ферментативної терапії. При хвороби Гоше необхідною умовою лікування вважається внутрішньоклітинний накопичення глікоцереброзідази, ймовірно, ефективним варіантом з'явиться пересадка селезінки хворому або створення можливості синтезу ферменту за допомогою специфічних фрагментів ДНК [Brady, 1966, 1969, Friedman, Robbin, 1972]. Аналогічний підхід може мати місце при хворобі Німана — Піка та інших Ліпоїдоз. При ідіотії Тея — Сакса лікування повинно проводитися до розвитку серйозних неврологічних порушень зазвичай у віці 2-6 міс. Доцільно використання переливань крові і плазми, тканинних екстрактів, антагоністів фолієвої кислоти і цитостатичних засобів, тиреоїдних гормонів, нікотинової кислоти та інших метаболічно активних сполук. За свідченнями необхідно проводити протисудомну і противоинфекционную терапію, застосовувати препарати, що зменшують спастику м'язів. Зазначені принципи можуть бути поширені на лікування хворих лейкодистрофії: противосудорожная і дегідратаційних терапія, застосування засобів, що регулюють м'язовий тонус і зменшують гіперкінези, препаратів амінокислот, вітамінів групи В і аскорбінової кислоти. При лікуванні хворих лейкодистрофії і внутрішньоклітинними Ліпоїдоз доцільно проводити повторні курси лікування в стаціонарі один — два рази на рік, а в інтервалах призначати амбулаторне лікування.
Збудником бруцельозу є мікроби роду Brucella. Природним резервуаром бруцел в природі є тварини, особливо кози і вівці, які представляють собою основне джерело інфекції для людини. Передача захворювання відбувається контактним, аліментарним або повітряним шляхами. Хворіють на бруцельоз переважно люди у віці 20-50 років, частіше уражаються особи, які мають контакт з тваринами (пастухи, ветеринари, працівники тваринницьких ферм, м'ясокомбінатів).
Патогенез бруцельозу
Збудник захворювання через слизові оболонки шлунково-кишкового тракту або через пошкоджену шкіру проникає в лімфатичні вузли, де розмножується. З регіонарних вузлів бруцелли по лімфатичних шляхах і кровоносні судини поширюються по всьому організму. Потрапляючи з крові в тканини лімфоїдної або гемопоетичних системи, бруцели утворюють метастатичні вогнища, з яких потім при загостреннях процесу можуть знову потрапляти в кров. Велику роль в патогенезі бруцельозу, крім безпосереднього впливу інфекційного агента, відіграють аутоімунні реакції, що розвиваються на тлі бруцельозній інфекції.
Клініка бруцельозу
Тривалість інкубаційного періоду коливається в межах 1-3 тижнів, але іноді може затягуватися до декількох місяців. Клінічна картина бруцельозухарактеризується поліморфно симптомів, їх різноманітним поєднанням і ремиттирующим плином. Розрізняють гострі, підгострі і хронічні форми захворювання. Початкова стадія гострої форми захворювання характеризується підйомом температури до високих цифр, ознобом, вираженою пітливістю, болями в суглобах, порушенням сну. Нерідко відзначається геморагічний ді «атез з петехиальной висипом і наполегливими носовими кровотечами. Поразка нервової системи зустрічається в основному при підгострому або хронічномубруцельозі, який може слідувати після гострої фази або розвиватися самостійно. Поразка периферичної нервової системи становить усіх форм Нейробруцельоз. Часто виникають дуже хворобливі невралгії, радикулярний синдром, що супроводжуються також артралгіями і міалгія. Ураження центральної нервової системи зустрічається рідше у вигляді енцефалітів, менінгоенцефалітів і менінгітів. Особливо часто виникають симптоми, які свідчать про поразку вегетативної нервової системи — загальна слабкість, підвищена стомлюваність, дратівливість, підвищена пітливість, хворобливість м'язів. Постійним симптомом захворювання є головний біль — інтенсивність її може широко варіювати — від незначно вираженої до дуже сильної. Змінюється поведінка хворих, з'являються скарги іпохондричного характеру. Іпохондрія супроводжується тривогою, нав'язливими думками, розгубленістю. Хворі починають прислухатися до своїх відчуттів, вважають себе тяжко хворими. У деяких випадках розвиваються психози.
Діагноз бруцельозу
Діагноз бруцельозу в зв'язку з великим поліморфізмом клінічних симптомів може представляти значні труднощі. Необхідно провести диференційний діагноз з туберкульозом, ревматизмом, туляремією, сепсисом, лімфогранулематозом, лейшманіоз і багатьма іншими захворюваннями. Діагноз бруцельозу обов'язково повинен бути підтверджений позитивними результатами лабораторних досліджень. Високо специфічною є реакція аглютинації Райта, що з'являється на 2-3-му тижні захворювання. У титрах 1: 400 і вище реакція Райта підтверджує діагноз бруцельозу. Прискорена реакція аглютинації Хедельсона більш чутлива, ніж реакція Райта, але менш специфічна. Алергічна проба Бюрне проявляється на 2-3-му місяці захворювання, вона підтверджує стан сенсибілізації але бруцельозного антигену, але не може служити показником ступеня активності захворювання . Для діагностики бруцельозу необхідно мати результати мінімум двох реакцій — реакції Райта і проби Бюрк.
Лікування бруцельозу
Для лікування гострих і підгострих форм бруцельозу застосовують антибіотики і лікувальні вакцини. З антибіотиків застосовують тетрациклін, левоміцетин, еритроміцин. Препарати приймають всередину протягом 5-7днів, потім слідує перерва 5 днів і знову повторюють 5-7-денний курс лікування. При необхідності призначають і третій, курс лікування. Дози тетрацикліну, еритроміцину 200 ТОВ ЕД 4 рази на добу. Левоміцетин приймають по 0,5 г в капсулах 4 рази на добу. Для лікування застосовують також стрептоміцин по 1 г на добу протягом 20 днів. протівобруцеллезного вакцину вводять внутрішньовенно. Вона особливо показана при хронічних формах бруцельозу. Застосування кортикостероїдів показано при гострому бруцельозі з тяжким перебігом та поганою переносимістю антибіотиків. У хворих невритами і радикуліту бруцельозній етіології преднізолон не ефективний [Беклемишев Н. Д., 1963].
Ці ускладнення обумовлені алергічною реакцією на мієлін мозкової тканини, що міститься у вакцині. Патологоанатомічно розвивається ускладнення розцінюється як гніздову дисемінований, іноді розлитої, енцефаломієліт і поліневрит з невеликою менінгеальної реакцією. Страждає переважно сіра речовина. У периферичних нервах, як і в мозку, судинні зміни переважають над дегенеративними. Перші ознаки ускладнення з'являються зазвичай після початкових сеансів, частіше після 4-8-й ін'єкції антирабічної вакцини. Продромальний період протікає в формі запаморочення, загальної слабкості, нудоти або проносів, болів в місці ін'єкції і у всьому тілі. Потім підвищується температура, з'являється головний біль, оглушення, блювота, часто неприборкана. Зіниці спочатку широкі, потім стають вузькими, часто виникає косоокість. Іноді втягується і лицьовий нерв. Можливі бульбарні явища: порушення ковтання, почастішання пульсу та дихання. Картина одночасного ураження периферичних нервів і спинного мозку розгортається і прогресує протягом 2-6 днів. Розвивається млявий параліч ніг або рук, з'являються сфінктерние порушення і рідше розлади чутливості по проводниковому або полиневритическому типу. У зонах порушеною іннервації кінцівок тканини болючі на дотик. У спинномозковій рідині часто знаходять підвищений вміст білка і невеликий плеопітоз з переважанням лімфоцитів. Регрес симптомів енцефаломіелорадікулоневрітов починається швидко -як зазвичай при алергічних процесах і лише в поодиноких спостереженнях розтягується на місяці зі збереженням резидуальних парезів і розладів чутливості. Тяжкість і поширеність процесу варіюють від блискавично протікає висхідного паралічу, який спливає в 1/3 випадків смертельно, до абортивних мононевритов.
Новини по темі:
[2015.11.02]
Протягом останніх десятиліть для людства ознаменувалися безліччю відкриттів в області медицини і зокрема інфекційних захворювань. У число цих відкриттів увійшли і численні штами грипу, які спалахують в різних країнах і регіонах. Визначившись з ворогами, вчені почали працювати над боротьбою з ними … масової боротьбою. Розвиток
Ця форма захворювання описана в 1886 р Eulenburg. У класичних випадках захворювання проявляється тільки на холоді, коли виникає Міотонічна утруднення розслаблення м'язів. Локальне охолодження, наприклад при їжі морозива, супроводжується м'язовим спазмом типу миотонической контрактури в глотці і мовою, який проходить після зігрівання. Поза охолодження у хворих відзначається лише підвищена механічна збудливість м'язів. У деяких випадках охолодження, крім Міотонічна феномена, призводить до розвитку значної м'язової слабкості (так звані холодові паралічі). Захворювання передається по аутосомно-домінантним типом, зустрічається дуже рідко. Є внутрісімейний поліморфізм: у деяких членів однієї сім'ї може бути тільки миотония, у інших тільки напади м'язової слабкості, в останніх випадках захворювання дуже нагадує сімейну епізодичну адинамию. З віком є тенденція до поліпшення стану. Парадоксальна миотония. Зустрічається вкрай рідко. Виявляється типовим міотоніческім спазмом, який на відміну від миотонии Томсена при повторних рухах не тільки не зменшується, але навпаки наростає і рухи стають неможливими. Синдром Шварца — Джампел. Описано в 1962 р Schwarz і Jampel, його синонімами є хондродістрофіческая мнотонія, остеохондромишечная дистрофія. Захворювання проявляється в ранньому дитячому віці, коли відзначають відставання в рості, ряд аномалій розвитку (коротка шия, деформація грудної клітки, сколіоз), а також виражене постійне тонічне напруження м'язів, часті хворобливі м'язові скорочення в різних м'язових групах типу поширених «крамп». Найбільш постійним синдромом в клінічній картині є миотонический феномен з характерним ускладненням розслаблення і підвищеною механічною збудливістю м'язів. Інші симптоми можуть іноді бути відсутнім. Інтелект не страждає. Захворювання відноситься до спадкових з аутосомно-рецесивним типом передачі, частіше спостерігається при кровноспоріднених шлюби. Є вказівка про дуже хорошому ефекті лікування діакарб [Авер'янов Ю. М., 1978].
Патоморфологня і патогенез миотонии
При патоморфологічної дослідженні виявляються в основному зміни в м'язовій тканині. Так, при миотонии Томсена виявляються деяка атрофія м'язових волокон і переміщення їх ядер в центр. При дистрофічних миотонии має місце атрофія м'язових волокон, розростання сполучної тканини. Ядра збільшуються в кількості і розташовуються під сарколеммой, а також в товщі м'язового волокна і утворюють довгі ланцюжки. При гістохімічному дослідженні у хворих з дистрофічній міотонію знаходять зменшення в розмірі і збільшення числа волокон I типу, в той час як волокна II типу (білі) не змінюються або кілька гіпертрофуються. При електронно-мікроскопічному дослідженні знаходять гіпертрофію саркотубулярной системи, збільшення розмірів мітохондрій, а при дистрофічних миотонии виявляється додатково виражена деструкція миофибриллярного апарату, руйнування мітохондрій, збільшення кількості лізосом. Патогенез міотоніческім порушень розслаблення м'язів пов'язують з порушенням проникності клітинних мембран, зміною іонного і медиаторного обміну. Різноманітні синдроми, що становлять клінічну картину атрофической миотонии, можуть певною мірою можна пояснити порушенням регуляції гіпоталамо-гіпофізарної системи або плейотропних дією мутантного гена. Питання про нозологічної самостійності миотонии Томсена і атрофической миотонии в даний час може вважатися вирішеним позитивно. Крім своєрідності клінічної картини, у хворих є різні патоморфологічні зміни і зміни деяких біохімічних показників. Так, при атрофической миотонии знаходять збільшений катаболізм імунних глобулінів класу G, а також встановлена група зчеплення з секретером. Останній: факт використовується для антенатальної діагностики атрофической миотонии за допомогою методу амніоцентеза.
Лікування миотонии
При миотонии Томсена хінін, новокаїнамід, препарати кальцію надають незначний ефект. АКТГ у великих дозах може ліквідувати міотоніческіе симптоми, але через багатьох побічних явищ, а також короткочасність його дії таке лікування не може бути рекомендовано. У ряді випадків (майже в 50%) задовільний і навіть хороший ефект отриманий при застосуванні дифенина (по 0,1 г 3-4 рази на день переривчастим курсом). Лікування при атрофической миотонии малоефективно. У початкових стадіях, коли ще виражений миотонический спазм, що утрудняє активні рухи, показаний прийом дифенина. Призначаються також препарати синтетичних андрогенів, проводиться загальнозміцнююча терапія. У розгорнутих стадіях хвороби слід призначати повторні курси анаболічних гормонів в поєднанні з дробовим переливанням крові або плазми, вітамін Е, комплекс вітамінів В, невеликі дози АТФ.