
 Синдром Ретта важка і рідкісна спадкова патологія, яка трапляється у дітей жіночої статі.
Синдром Ретта важка і рідкісна спадкова патологія, яка трапляється у дітей жіночої статі.
Захворювання описано австрійським лікарем Андреасом Реттом, який першим помітив схожі симптоми у кількох дівчаток і припустив окреме психоневрологічне захворювання. Вчений досліджував і описав 31 випадок патологічних порушень.
В даний час встановлено, що синдром Ретта пов'язаний з мутацією Х-хромосоми. Оскільки у дівчаток є дві хромосоми, вони доживають до кінця гестаційного періоду і здаються на перший погляд цілком здоровими. Хлопчики, які мають одну Х-хромосому, виявляються нежиттєздатними і гинуть у внутрішньоутробному періоді або в перші місяці життя.
Класичними проявами синдрому Ретта є стереотипні руху рук, як ніби пацієнтка постійно їх миє, а також заламування і стискання кистей .
Подібні симптоми можна сплутати з проявами аутизму, але подальша клінічна картина обох захворювань дещо відрізняється. Дегенеративні зміни головного мозку прогресують і в майбутньому пацієнти втрачають набуті моторні та психічні навички.
Зміст
Статистичні дані
Згідно статистикою, захворювання схильна до 1 дівчинка на 10000-15000 новонароджених немовлят жіночої статі. Найбільш часто синдром Ретта зустрічається в Норвегії, Італії, Угорщини. Відомі спорадичні випадки розвитку синдрому в окремих сільських районах.
Спадкові аномалії були виявлені у однояйцевих близнюків і тих пар близнюків, які виховувалися в різних соціальних середовищах.
Асоціація синдрому Ретта
Пацієнти з рідкісним захворюванням постійно потребують певного догляду і адекватному сприйнятті їх рухових і  інтелектуальних функцій оточуючими людьми.
інтелектуальних функцій оточуючими людьми.
Тому в Росії існує Асоціація синдрому Ретта, головним завданням якої є медичне консультування батьків, інформаційна підтримка і допомога в реабілітації.
На є форма зворотного зв'язку, де батьки можуть задати свої питання, а також ознайомитися з новітніми досягненнями світової медицини з лікування захворювання.
Світовий конгрес
Світовий конгрес по синдрому Ретта відбувався в травні 2016 року в Казані, його метою було об'єднати вчених, практичних лікарів і батьків пацієнтів, які страждають рідкісним недугою.
На конгресі були присутні медичні фахівці з Італії, Нідерландів, Ізраїлю, Японії, США і Росії.
Науковий комітет прийшов до висновку, що педіатри не дуже орієнтуються в перших проявах синдрому, причому багато лікарів приділяють мало уваги реабілітації пацієнтів з рідкісними (Орфа) захворюваннями. Організаторами заходу стали: Міністерство охорони здоров'я Росії, Татарстану, некомерційні об'єднання.
На конференції були озвучені новітні досягнення вчених і визначені подальші шляхи лікування недуги. Також був присутній головний позаштатний спеціаліст з генетичним дослідженням РФ Сергій Куцев, який зазначив, що в Росії діти з рідкісними захворюваннями перебувають поза зоною уваги фахівців соціальних служб, багатьом з них недоступні реабілітаційні центри.

Генні мутації визначальний фактор
Фахівці вважають, що причина криється в генетичної мутації, пов'язаної з геном MECP2. В результаті спадкового порушення виявляється дефіцит глутамінової рецепторів в базальних гангліях, рецепторів дофаміна в хвостатому ядрі, порушення холінергічних структур. Ген MECP2 асоційований з Х-хромосомою і контролює особливий білок (МЕСР2).
При нормальній функції гена цього білка забезпечує правильний розвиток головного мозку, але якщо є мутація, відбувається регрес розумових і фізичних навичок. Уповільнення відставання мозкових структур виявляється до 4-річного віку дитини.
Перші ознаки
 Дівчатка народжуються без особливих відхилень і педіатри не знаходять аномалій аж до 6-18 місяців. Єдине, що може насторожити лікаря — це низька температура тіла і гіпотонія м'язів тулуба. Такі ознаки нерідко списують на післяпологові порушення, тому мало приділяється уваги діагностиці та лікуванню.
Дівчатка народжуються без особливих відхилень і педіатри не знаходять аномалій аж до 6-18 місяців. Єдине, що може насторожити лікаря — це низька температура тіла і гіпотонія м'язів тулуба. Такі ознаки нерідко списують на післяпологові порушення, тому мало приділяється уваги діагностиці та лікуванню.
Дітей направляють до невропатолога, який виявляє уповільнення темпів фізичного розвитку: пізніше перевертання немовляти на спину, відсутність навичок повзання.
При огляді визначають надлишкову пітливість долонь, блідість шкіри. Надалі рухові порушення прогресують.
Стадії розвитку
Відомі 4 стадії розвитку хвороби:
- Стагнація — тимчасове призупинення хвороби, при якій не відбувається наростання симптомів. Триває від 6-18 місяців і більше. Дитина втрачає інтерес до навколишніх подій, помітна гіпотонія м'язів, уповільнення росту голови і кінцівок.
- Погіршення стану . Стадія триває від 1 року до 3-4 років. Якщо дитина опанував навичками мови, пересування, вони поступово зникають. З'являються стереотипні маніпуляції руками, порушення з боку легеневої системи (гіпервентілляція, задишка, раптова зупинка дихання), дискоординація рухів, невмотивоване занепокоєння. Вже на 2 стадії з'являються судомні напади, лікування яких не є результативним.
- Відносна стабільність , ця стадія може тривати аж до раннього шкільного віку. Відзначається розумова недостатність, судоми, занадто мало ваги, порушення емоційного контакту з оточуючими. змінюються загальмованістю нервової системи.
- Завершальна стадія характеризується зниженням частоти судом, зате з'являється кахексія, сколіоз, виражені порушення з боку дихання. Можлива неспроможність пересування, визначають низький зріст кінцівок і малу окружність голови.
Характеристика порушень
Багато в чому діти з синдромом Ретта нагадують пацієнтів з аутизмом, на це вказують такі симптоми:
- відсутність емоційного і зорового контакту з оточуючими:
- однотипні руху рук і тіла;
- втрата соціальних навичок;
- плач, спонтанний крик, неспокійна поведінка без видимої причини;
- порушення чутливості;
- нетримання калу і сечі;
- прогресування симптомів.

Однак варто звертати увагу і на інші ознаки, характерні для синдрому Ретта, а саме:
- легеневу патологію;
- відставання в рості кистей і стоп;
- прогресуючу дискоординацию рухів;
- порушення з боку рухового апарату (, сколіоз) ;
- часті , резистентні до терапії протисудомними засобами;
- мікроцефалію (малий череп);
- кахексию (в пізніх стадіях) .
Поступово обличчя дитини втрачає колишню вираз і більше нагадує маску, погляд перестає бути цілеспрямованим, блукає або спрямований в одну точку.
Відео-сюжет про синдром Ретта:
Сучасна діагностика
діагностика захворювання включає методи: 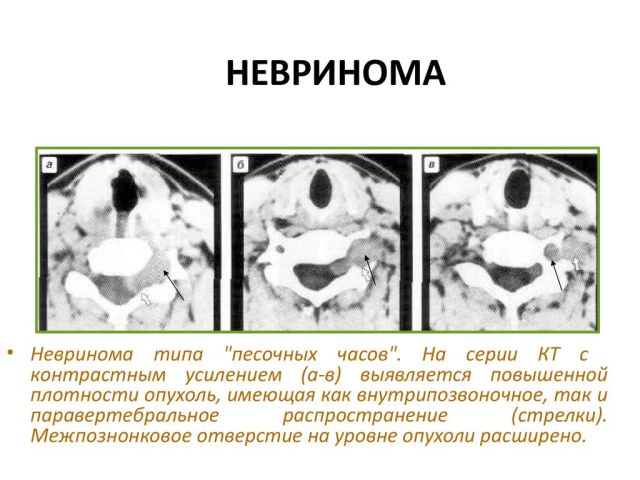
- електроенцефалографію;
- ;
- ЕКГ (визначається подовження інтервалу QT, періодичне уповільнення ритму);
- молекулярно-цитологічну діагностику .
Обов'язково враховуються клінічні ознаки, при типових проявах призначається дослідження мутації генів на Х-хромосомі.
Труднощі полягають в малій обізнаності лікарів про рідкісних станах, часом доводиться проходити безліч аналізів і відвідувати велике число фахівців, поки діагноз не буде встановлений. Крім того, складні генетичні дослідження доступні тільки у великих містах, де є наукові центри.
Лабораторними дослідженнями визначається:
- підвищення глутамату і бета-ендорфінів;
- атипові гліколіпіди в головному мозку;
- зниження субстанції P;
- зменшення нервових чинників зростання.
На що здатна сучасна медицина?
З медикаментів призначаються антиконвульсанти, антипсихотичні препарати. Застосовуються засоби для поліпшення кровообігу і стимуляції роботи головного мозку. Лікування в основному симптоматичне.
 Велике значення має навчання батьків способам спілкування з дитиною, трудотерапія, посилення комунікаційних навичок. Особлива увага приділяється іграм, як способу розвитку мовних, моторних і когнітивних функцій.
Велике значення має навчання батьків способам спілкування з дитиною, трудотерапія, посилення комунікаційних навичок. Особлива увага приділяється іграм, як способу розвитку мовних, моторних і когнітивних функцій.
Проводиться лікування сколіозу. Такі діти потребують регулярних заняттях лікувальною фізкультурою для відновлення м'язового тонусу і масажі.
При шлунково-кишкових розладах контролюють частоту стільця, призначаються проносні засоби і дієта з підвищеним вмістом клітковини. Для запобігання кахексії рекомендується калорійне харчування кожні 3 години.
Прогнози і очікування
Останні дослідження показали, що відновлення функції гена MECP2 дозволяє усунути неврологічні порушення. Наукові дослідження проводилися на лабораторних мишах, в ході експериментів було доведено оборотність психічних і нервових порушень, відбувається згасання ознак хвороби.
Ці експерименти дають надію пацієнтам та їхнім батькам на те, що моторні, мовні та дихальні функції можуть бути відновлені .
Статистика показує, що жінки з синдромом Ретта можуть прожити 40 і більше років. Основні причини летальності: зупинка серця, судоми, прорив стінки шлунка, дисфункція стовбура головного мозку.
 Під Парасомнии (грец. Para — біля, проти + лат. Somnus — сон) розуміють цілий ряд розладів сну, що характеризуються поведінковими, руховими або вегетативними феноменами. Говорячи по-іншому, неприродним, дивною поведінкою людини під час сну.
Під Парасомнии (грец. Para — біля, проти + лат. Somnus — сон) розуміють цілий ряд розладів сну, що характеризуються поведінковими, руховими або вегетативними феноменами. Говорячи по-іншому, неприродним, дивною поведінкою людини під час сну.  плачем.
плачем.  Лунатизм? найдивніша і складна форма парасомніі, недарма він обріс такою кількістю міфів. Зазвичай сомнамбулізм відбувається в фазі глибокого сну в 1-ій половині ночі, але іноді трапляється і під час швидкого сну під ранок.
Лунатизм? найдивніша і складна форма парасомніі, недарма він обріс такою кількістю міфів. Зазвичай сомнамбулізм відбувається в фазі глибокого сну в 1-ій половині ночі, але іноді трапляється і під час швидкого сну під ранок.  криком, плачем. При цьому повного пробудження не відбувається. Стан небезпечно тим, що можуть наноситися травми? як самому собі, так і оточуючим.
криком, плачем. При цьому повного пробудження не відбувається. Стан небезпечно тим, що можуть наноситися травми? як самому собі, так і оточуючим. Мовлення уві сні зазвичай триває недовго і складається як з простих коротких звуків, так і довгих речень. Виявляється під час переходу зі сну в пильнування.
Мовлення уві сні зазвичай триває недовго і складається як з простих коротких звуків, так і довгих речень. Виявляється під час переходу зі сну в пильнування. 
 В цілому прогноз по парасомнии вважається сприятливим,? за винятком важких невралгічних захворювань і психосоматичних розладів. При своєчасному зверненні до лікаря, вони не тягнуть небезпечних для життя наслідків.
В цілому прогноз по парасомнии вважається сприятливим,? за винятком важких невралгічних захворювань і психосоматичних розладів. При своєчасному зверненні до лікаря, вони не тягнуть небезпечних для життя наслідків. 
 браділалія це порушення темпо ритмічної сторони мовлення, пов'язане зі стійкими труднощами при відтворенні розчленованих звуків. Ця патологія відноситься до категорії розладів темпо-ритмічної сторони мовлення несудорожним характеру. У важких випадках може виникати заїкання.
браділалія це порушення темпо ритмічної сторони мовлення, пов'язане зі стійкими труднощами при відтворенні розчленованих звуків. Ця патологія відноситься до категорії розладів темпо-ритмічної сторони мовлення несудорожним характеру. У важких випадках може виникати заїкання. 


 Крім загального обстеження мови проводиться аналіз загальної та ручної моторики, мімічних реакцій, сенсорних функцій, рівня інтелекту.
Крім загального обстеження мови проводиться аналіз загальної та ручної моторики, мімічних реакцій, сенсорних функцій, рівня інтелекту.  задає логопед і підтримує за допомогою диригування і ударів. Надалі вправи поступово ускладнюються від промовляння складів, слів, пропозицій до читання тексту в ролях, декламування віршів і скоромовок.
задає логопед і підтримує за допомогою диригування і ударів. Надалі вправи поступово ускладнюються від промовляння складів, слів, пропозицій до читання тексту в ролях, декламування віршів і скоромовок.  гіперактивними дітьми . Ні в якому разі не можна лякати дитину злими і страшними персонажами.
гіперактивними дітьми . Ні в якому разі не можна лякати дитину злими і страшними персонажами. СМА або спінальна м'язова атрофія — це група з декількох видів спадкових патологій, в результаті яких відбувається часткова або повна втрата рухових нейронів спинного мозку в передній частині.
СМА або спінальна м'язова атрофія — це група з декількох видів спадкових патологій, в результаті яких відбувається часткова або повна втрата рухових нейронів спинного мозку в передній частині.  груп, периферичних нервів і спинного мозку, при цьому він зазначив атрофію клітин передніх рогів і корінців спинного мозку.
груп, периферичних нервів і спинного мозку, при цьому він зазначив атрофію клітин передніх рогів і корінців спинного мозку.  Хвороба помітна ще з перших годин народження, відсутністю глибоких рефлексів і вираженою м'язовою гіпотонією. Яскраво виражені кістково-суглобові деформації. Навіть у тієї невеликої кількості діток, які зможуть сісти і тримати голову ці навички скоро регресують.
Хвороба помітна ще з перших годин народження, відсутністю глибоких рефлексів і вираженою м'язовою гіпотонією. Яскраво виражені кістково-суглобові деформації. Навіть у тієї невеликої кількості діток, які зможуть сісти і тримати голову ці навички скоро регресують. 

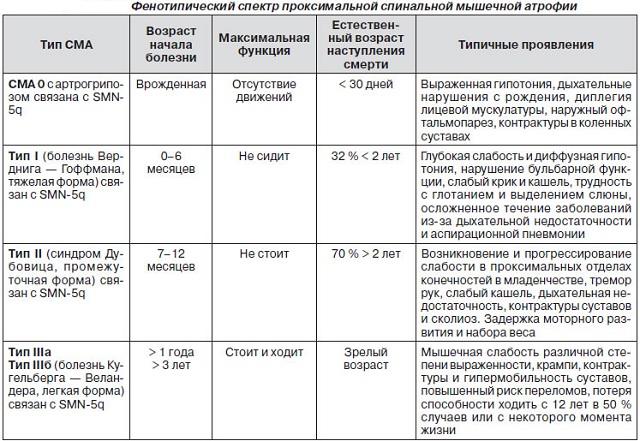
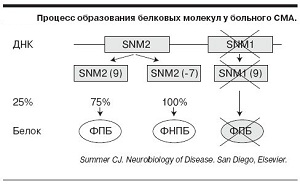 Так як дане захворювання відбувається здебільшого з -за нестачі SMN-білка, основне завдання лікування полягає в підвищенні його рівня. На сьогодні в цьому напрямку ведуться активні дослідження в інститутах США, Італії, Німеччини.
Так як дане захворювання відбувається здебільшого з -за нестачі SMN-білка, основне завдання лікування полягає в підвищенні його рівня. На сьогодні в цьому напрямку ведуться активні дослідження в інститутах США, Італії, Німеччини. 
 Аненцефалія це патологія внутрішньоутробного розвитку мозку, яка виникає на тлі несмиканія нервової трубки.
Аненцефалія це патологія внутрішньоутробного розвитку мозку, яка виникає на тлі несмиканія нервової трубки. 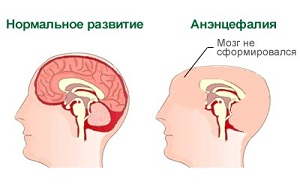 може дефіцит в організмі матері фолієвої кислоти.
може дефіцит в організмі матері фолієвої кислоти.

 необхідні вітаміни не тільки під час виношування дитини, але і при плануванні вагітності. Особливо важливу роль відіграє фолієва кислота, дефіцит якої в організмі жінки може спровокувати внутрішньоутробні вади нервової трубки. До застосування показано 400 мікрограмів кислоти на добу.
необхідні вітаміни не тільки під час виношування дитини, але і при плануванні вагітності. Особливо важливу роль відіграє фолієва кислота, дефіцит якої в організмі жінки може спровокувати внутрішньоутробні вади нервової трубки. До застосування показано 400 мікрограмів кислоти на добу. Дисграфія це повне або часткове порушення письмовій діяльності, яка пов'язана з дефіцитом сформованості тих функцій психіки, які відповідальні за виконання і контроль листи.
Дисграфія це повне або часткове порушення письмовій діяльності, яка пов'язана з дефіцитом сформованості тих функцій психіки, які відповідальні за виконання і контроль листи. 




 Діти, які страждають таким порушенням, вимагають кваліфікованої логопедичної допомоги, т. к. усунути цю проблему в звичайних шкільних умовах просто неможливо.
Діти, які страждають таким порушенням, вимагають кваліфікованої логопедичної допомоги, т. к. усунути цю проблему в звичайних шкільних умовах просто неможливо. 


 мієлітів називають досить рідко зустрічається вид запалення спинного мозку. Хвороба поширюється або по всьому хребту, або може зачіпати окремі його частини.
мієлітів називають досить рідко зустрічається вид запалення спинного мозку. Хвороба поширюється або по всьому хребту, або може зачіпати окремі його частини.  ураження відділів спинного мозку. Зустрічаються і проблеми з заднім канатиком, передніми шляхами і діаметром. Дізестезія спостерігається в одній нозі, а потім піднімається по обох ногах.
ураження відділів спинного мозку. Зустрічаються і проблеми з заднім канатиком, передніми шляхами і діаметром. Дізестезія спостерігається в одній нозі, а потім піднімається по обох ногах. Разом з нездужанням хворі відчувають біль в спині. Всі симптоми хвороби залежать від тяжкості перебігу, а це говорить про те, що зміни відбуваються нижче межі вогнища.
Разом з нездужанням хворі відчувають біль в спині. Всі симптоми хвороби залежать від тяжкості перебігу, а це говорить про те, що зміни відбуваються нижче межі вогнища. 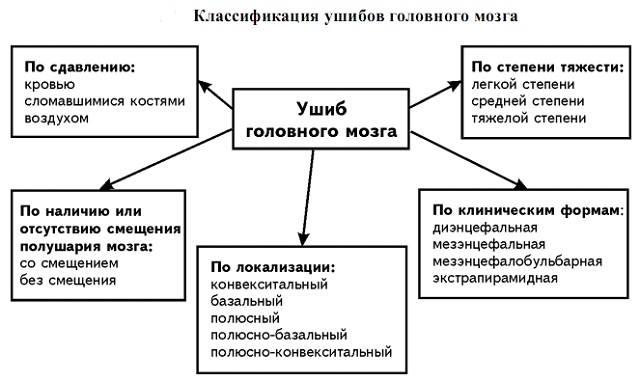
 підозра на бактеріальний характер хвороби або для лікування септичного ускладнення.
підозра на бактеріальний характер хвороби або для лікування септичного ускладнення.  очищену поверхню накладати мазь Вишневського або масло обліпихи. Для якнайшвидшого загоєння пролежнів можна промивати їх свіжовичавленим помідоровим соком або інсуліну.
очищену поверхню накладати мазь Вишневського або масло обліпихи. Для якнайшвидшого загоєння пролежнів можна промивати їх свіжовичавленим помідоровим соком або інсуліну. 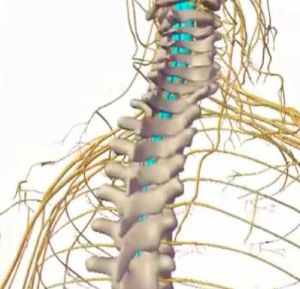
 Під краніостеноз розуміють передчасне зрощення черепних швів, що сприяє обмеженню загального обсягу черепа, його деформації і провокує розвиток .
Під краніостеноз розуміють передчасне зрощення черепних швів, що сприяє обмеженню загального обсягу черепа, його деформації і провокує розвиток . 
 Для неї характерно: подовження і звуження черепа, розширення чола, крайнє зрощення або закриття великого джерельця. При даній формі патології природні пологи часто ускладнені, що пов'язано з невідповідністю розмірів голівки плоду і родових шляхів матері.
Для неї характерно: подовження і звуження черепа, розширення чола, крайнє зрощення або закриття великого джерельця. При даній формі патології природні пологи часто ускладнені, що пов'язано з невідповідністю розмірів голівки плоду і родових шляхів матері.
 наявність пальцевих вдавлений на черепних кістках, деформацію черепа;
наявність пальцевих вдавлений на черепних кістках, деформацію черепа;  Існує величезна кількостей оперативних методик в залежності від типу краніостеноз — краніотомія лінійна, клаптева двостороння, циркулярна, резекція зводу черепа та інші.
Існує величезна кількостей оперативних методик в залежності від типу краніостеноз — краніотомія лінійна, клаптева двостороння, циркулярна, резекція зводу черепа та інші. 
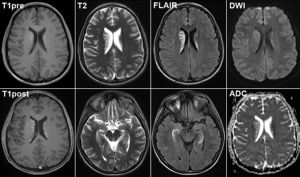
 Енцефаліт — запалення головного мозку, причиною якого є проникнення різних мікроорганізмів в нього. Розрізняють первинний і вторинний .
Енцефаліт — запалення головного мозку, причиною якого є проникнення різних мікроорганізмів в нього. Розрізняють первинний і вторинний .  сільській місцевості ризикують стати жертвами комах, укуси яких і викликають запальні процеси головного мозку.
сільській місцевості ризикують стати жертвами комах, укуси яких і викликають запальні процеси головного мозку. Після потрапляння вірусу і проходження інкубаційного періоду починають поступово проявлятися симптоми. Що характерно, не обов'язково будуть присутні всі одночасно — це залежить від кожного індивідуального випадку, деякі можуть не з'явиться зовсім.
Після потрапляння вірусу і проходження інкубаційного періоду починають поступово проявлятися симптоми. Що характерно, не обов'язково будуть присутні всі одночасно — це залежить від кожного індивідуального випадку, деякі можуть не з'явиться зовсім.  Енцефаліт в легкій формі можна вилікувати приблизно за три місяці. Що ж стосується важких форм — цей процес може затягнутися на довгі роки. Що характерно, при тяжкому перебігу хвороби летальний результат наступає приблизно в 70% випадків .
Енцефаліт в легкій формі можна вилікувати приблизно за три місяці. Що ж стосується важких форм — цей процес може затягнутися на довгі роки. Що характерно, при тяжкому перебігу хвороби летальний результат наступає приблизно в 70% випадків .  слинотеча, підвищена пітливість, сальність особи.
слинотеча, підвищена пітливість, сальність особи.  Найбільш небезпечною є блискавична форма енцефаліту: всі симптоми розвиваються гранично швидко: різкий стрибок температури тіла, сильний головний біль, практично моментальне порушення свідомості і кома.
Найбільш небезпечною є блискавична форма енцефаліту: всі симптоми розвиваються гранично швидко: різкий стрибок температури тіла, сильний головний біль, практично моментальне порушення свідомості і кома.