Закриті пошкодження хребта і спинного мозку
пошкодження хребта і спинного мозку поділяються на закриті — без порушення цілості шкірних покривів і підлягають м'яких тканин, відкриті -з порушенням цілості останніх (вогнепальні і колото-різані поранення).
Закриті пошкодження хребта в свою чергу діляться на дві групи:
- Неускладнені пошкодження хребта без порушення функції спинного мозку або його корінців.
- Ускладнені ушкодження хребта з порушенням функції спинного мозку і його корінців:
- з рентгенологічно виявляються переломами, переломовивіхах, вивихами тіл хребців,
- без рентгенологічно виявляються пошкоджень хребта.
У мирний час частота пошкоджень спинного мозку і його корінців при закритих травмах хребта становить близько 30% випадків. Переломи хребта з пошкодженням спинного мозку найчастіше зустрічаються в гірській промисловості, на транспорті, рідше на виробництві, в побуті, при спортивних вправах (особливо при пірнанні).
Найчастіше переломи хребта виникають в області Thxn-Ln, що пояснюється переважною передачею кінетичних сил на область з'єднання рухомих відділів хребта з відносно малорухомими. На другому місці за частотою стоять переломи, що локалізуються в області Cv-СVII т. Е. В області рухомих відділів шиї на кордоні з малорухливим грудним відділом.
Звертає особливу увагу досить часто зустрічається невідповідність між рентгенологічної картиною зміщення кісток і вагою неврологічної патології. При значно вираженій картині перелому і зміщення хребців може бути відсутнім клініка ураження спинного мозку, або вона виражена в незначній мірі і, навпаки, при відсутності рентгенологічних доказів компресії мозку можуть виникати різні симптоми ураження спинного мозку аж до синдрому повного поперечного перерви.
Види травм хребта і спинного мозку
Все порушення цілісності та функціональності хребта і спинного мозку поділяють на відкриті і закриті. Тобто супроводжуються ушкодженнями м'яких тканин і шкірних покривів і не відмічені такими, відповідно. Перші створюють додаткову небезпеку у вигляді ймовірності виникнення інфекції спинного мозку. Крім цього, розрізняють відкриті проникаючі травми, які характеризуються ушкодженнями не тільки м'яких тканин, але і твердої оболонки мозку. Закриті ж травми можуть призводити до порушень функціонування спинного мозку і корінців (ускладнені) або ж не супроводжуватися подібними ускладненнями.
Класифікація травм можлива згідно причин (згинання, удар і ін.), Характеру ( забій, перелом, вивих і т.д.). Важлива роль відводиться також відмінностей травм по їх стабільності, тобто ймовірності виникнення зсуву і його подальшого повтору. Крім того, види ушкоджень розрізняються по локалізації таких в різних відділах хребта.
Травма шийного відділу хребта і спинного мозку
Травма шийного відділу хребта несе найбільшу загрозу життю і здоров'ю хворого. У разі пошкодження спинного мозку ймовірність смерті вкрай велика унаслідок зупинки дихання, наступного за паралічем діафрагми. Частіше інших такі пошкодження (навіть без порушення цілісності спинного мозку) призводять до обмеженості опорно-рухової функції і сильних больових відчуттів, у разі впливу на спинний мозок, висока ймовірність втрати чутливості. Небезпеку становить і оперативне втручання в даному відділі, тому рішення про необхідність такого приймається в ситуації, коли ризик виправданий врятуванням життя або ж знижений загальними факторами.
Травма поперекового відділу хребта і спинного мозку
[ Рис.1 ] [ Рис.2 ]
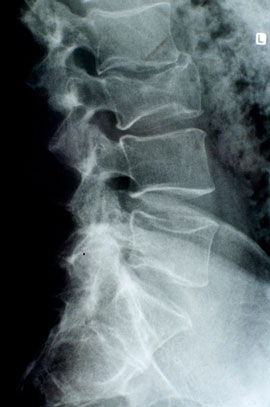
[Рис. 1] Рентген.
Зламаний поперековий хребець
Найбільш часто в клінічній практиці зустрічається травма поперекового відділу, оскільки дана локалізація відчуває максимальне навантаження при згинанні і розгинанні, підйомі тягарів тощо. Як правило, травма доводиться на верхню, малорухливу частина, в області I-III хребців. Дана локалізація ураження характеризується періодичними або постійними гострими болями, обмеженістю рухів при поворотах і згинанні тіла. Нерідко супроводжується порушенням роботи шлунково-кишкового тракту, парезом кишечника і затримками в роботі сечового міхура, здуттям живота і блювотою. Можливо порушення рефлекторної діяльності. Імовірність втрати чутливості досить висока. Високу ефективність в разі поразки поперекового відділу має реабілітація з урахуванням теплових процедур, ЛФК та масажу. Нерідко пацієнтам рекомендується пастельний режим тривалість до двох місяців. При компресії структури нерва або спинного мозку показано оперативне втручання.
Травми грудного відділу хребта і спинного мозку
Слід зазначити, що грудний відділ хребта малорухомий і більшою мірою стабільний. Однак при цьому він обмежений мобільним шийним і поперековим відділом, крім того в силу структури організму людини, ця частина хребта має вузький хребетним каналом. Нерідко ці факти стають вирішальними при отриманні травми, оскільки зумовлюють ускладнення. Найчастіше травми грудного відділу є удари або горизонтальні переломи, клиновидні деформації. Рідше зустрічаються осколкові і компресійні переломи. Як правило, методи лікування консервативні. Хірургічне втручання застосовується в разі ускладненої травми. У всіх випадках рекомендований досить тривалий постільний режим з мінімізацією вертикальних навантажень. Після лікування обов'язкові реабілітаційні заходи, що включають в себе ЛФК.
Симптоми травм хребта і спинного мозку
Залежно від ступеня складності травми, симптоми такої різняться. Зокрема, забій хребта виражається в хворобливості, припухлості ураженого місця. Біль, як правило, 'розливається', але може посилюватися до гострої, руху частково обмежені, приносять хворобливі, неприємні відчуття. Рідше зустрічаються підшкірні крововиливи, які супроводжують травму. При пальпації виявляється болючість. Анамнез зазвичай містить підняття важких предметів, різке скорочення м'язів, удар тощо.
При переломах і вивихах виникають локальні больові відчуття, біль може 'віддавати' в протилежну або хвору сторону, 'розливатися'. У разі порушення цілісності поперечних відростків, проявляє себе симптом Пайра і / або прилип п'яти. Травм хлистів призводять до болів в шийному відділі і голові, нерідко спостерігається оніміння кінцівок, порушення невралгії, функцій пам'яті. Трансдентальний вивих атланта нерідко виявляється причиною летального результату в зв'язку з різким впливом на довгастий мозок. В інших випадках положення голови може бути фіксованим або нестабільним, проявляється біль, нерідко повна або часткова втрата чутливості в ділянці шиї, неврологічна симптоматика.
[ Рис.1 ] [ Рис.2 ]
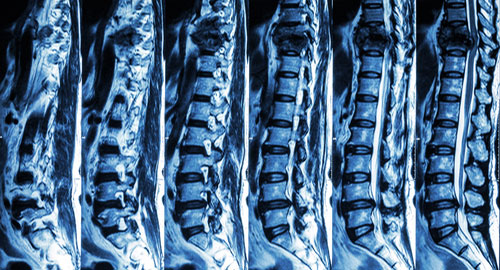
[Рис. 2] МРТ поперекового і грудного відділу хребта.
перелом грудного відділу хребта і стиснення спинного мозку
Пошкодження спинного мозку також виражається в залежності від критичності рівня. Найбільш критична область — рівень IV хребця шийного відділу. Травма, яка припала вище нього, призводить до паралічу діафрагми, що в свою чергу веде до повної зупинки дихання і летального результату. У всіх інших випадках прояви можуть полягати в порушенні або повній відсутності чутливості, обмеженої функціональності органів таза. У різних випадках може виникати сильна пекучий біль, часткова або повна втрата рухової функції, порушення рефлекторної активності, спазмування. Ускладнений дихання, кашель з виділенням легеневої секреції також є симптомами травми спинного мозку. Негативно позначається також сама по собі і на сексуальної функції. Кровотік і лімфоток також можуть сповільнюватися, приводячи до швидкого формування пролежнів. Для розриву спинного мозку характерно виразка шлунково-кишкового тракту з рясною кровотечею.
Морфологічні зміни спинного мозку при закритих пошкодженнях хребта
При закритій травмі хребта спостерігаються різні ступені пошкодження спинного мозку — від мікроскопічних до ударів, розтрощення і анатомічного перерви відповідно до рівня перелому і вивиху хребта. Набряк мозку може досягти такого ступеня, що мозок заповнює весь просвіт дурального каналу. При патологоанатомічному дослідженні у випадках смерті після закритих ушкоджень хребта з клінічними проявами ураження спинного мозку виявляються ураження нейронних структур у вигляді хроматолізіса (яке розцінюється як морфологічний прояв спинального шоку), осередки некрозу і розм'якшення, набухання і нерегулярність будови аксонів, дегенерація мієлінових обкладок, мелкоточечние, центральні гематомієлії, іноді інтра- і екстрадуральние геморагії, набряк спинного мозку, пошкодження корінців.
Внаслідок безпосереднього пошкодження молекулярних структур, розладів кровопостачання і кисневого голодування, пошкоджень судин і тканини спинного мозку, перифокального набряку, порушення ликвороциркуляции в спинному мозку можуть розвинутися некрози, розм'якшення, дегенеративні зміни клітинних і проводять структур і судинної системи, процеси організації і рубцювання, що супроводжуються патологічними змінами в оболонках, що клінічно проявляється різними синдромами.
Неврологічні симптоми при пошкодженнях хребта
Переломи хребта без порушень функцій спинного мозку зустрічаються частіше переломів з розладом цих функцій. Такі переломи не є небезпечними для життя, і при правильному лікуванні часто спостерігається повне одужання. Переломи хребта в поєднанні з пошкодженням спинного мозку є одними з найбільш прогностично несприятливих пошкоджень. Частота ускладнених переломів хребта становить близько 25% всіх переломів і залежить від характеру і локалізації пошкодження, а також умов його виникнення.
При всіх видах ушкодження хребта можуть виникати все ступеня пошкодження спинного мозку — від найлегших до незворотного синдрому поперечного ураження. При ускладнених пошкодженнях хребта синдром повного поперечного ураження спинного мозку зустрічається приблизно у 50% постраждалих.
Існують наступні синдроми травматичного ураження спинного мозку:
- струс
- забій (контузія спинного мозку)
- розтрощення
Під терміном « струс спинного мозку » (commotio spinalis) розуміють оборотне порушення його функцій при відсутності видимих пошкоджень структури мозку. Передбачається, що симптоми струсу спинного мозку є наслідком порушення функцій нервових клітин при раптовому виключенні супраспінальних впливів, а також мікроструктурних змін і парабиотического стану нервових клітин і нервових волокон нижче рівня ушкодження. При легких формах струсу зворотний розвиток симптомів відбувається в найближчі години після травми, при більш важких — в найближчі дні або тижні (до місяця).
У клінічній практиці початковий період травми, характеризується раптовим випаданням рухової, чутливої і рефлекторної діяльності, позначається терміном « спинальний шок ». Тривалість цього періоду в випадках оборотності неврологічної симптоматики дуже варіабельна і може досягати декількох тижнів і навіть місяців.
Під терміном « контузія спинного мозку » (contusio spinalis) розуміють забій його з пошкодженням самої тканини. При цьому в кінцевій стадії захворювання можуть спостерігатися залишкові явища порушення функції мозку. Забій спинного мозку в більшості випадків супроводжується картиною спинального шоку, т. Е. Тимчасовими парезами, паралічами, гіпотонією, арефлексією, розладами чутливості, порушенням функції тазових органів і деяких вегетативних функцій (потовиділення, піломоторних рефлексів, помилкової температури і т. Д.). Симптоми спинального шоку затушовують справжню картину пошкодження спинного мозку, і тільки коли мине ознак шоку залишається стійка симптоматика, що є наслідком удару мозку або його розтрощення.
У більшості випадків картина ураження спинного мозку досягає піка відразу ж після травми хребта, що свідчить про значення раптової зміни конфігурації хребетного каналу на рівні ушкодження. Тільки в порівняно рідкісних випадках в наступний період спостерігається прогресування неврологічних симптомів в результаті набряку і крововиливів. При неврологічному обстеженні в найближчі години після травми необхідно в першу чергу з'ясувати, чи є картина повного поперечного ураження спинного мозку або тільки часткове випадання його функцій. Збереження будь-яких елементів моторики або чутливості нижче рівня ушкодження свідчать про часткове ураженні спинного мозку. Тривало існуючий приапизм і ранні трофічні розлади, як правило, свідчать про необоротний пошкодженні мозку. Якщо в клінічній картині повного поперечного ураження в найближчі 24-48 год може залишитись непоміченим ознак відновлення функцій, то це зазвичай свідчить про незворотність пошкодження і є поганим прогностичним ознакою.
Симптоми ураження спинного мозку при травмі хребта відображають різні фази захворювання. Спочатку з'являються ознаки спинального шоку у вигляді раптово розвинулася млявою параплегії, відсутність чутливості, арефлексии нижче рівня ураження, затримки сечовипускання і дефекації, нерідко з приапизмом і відсутністю потовиділення нижче рівня ураження.
Гістологічно ця фаза проявляється хроматолізісом уражених нейронів. Потім наростає спінальна рефлекторна активність з виникненням спастичних явищ, спинального автоматизму і в ряді випадків сгибательного спазму. Відновлення рефлекторної активності починається значно дистальніше рівня ураження, піднімаючись вище аж до цього рівня.
Однак при розвитку важкого урогенная сепсису, бронхопневмонії або інтоксикації внаслідок пролежнів стадія спінальної рефлекторної активності може знову змінитися млявою параплегией і арефлексією, що нагадують стадію спинального шоку.
гематомієлія. у випадках локалізації гематомієлії в шийному відділі часто спостерігаються летальні випадки. У патогенезі дихальних порушень при пошкодженні на рівні Civ-Cv шийного сегмента має значення розвивається при цьому параліч діафрагми. При наявності спинального шоку його симптоми затушовують картину гематомієлії, і вона може клінічно проявитися значно пізніше.
Синдром ураження передніх відділів спинного мозку. Синдром ураження передньої спінальної артерії, описаний в основному при судинних ураженнях спинного мозку, може спостерігатися і при травматичному його поразці, оскільки передня спинальна артерія постачає 2/3 речовини спинного мозку. Для цього синдрому характерні паралічі з диссоційованними розладами чутливості і порушення функцій тазових органів, але за відсутності ознак ураження задніх стовпів.
Синдром ушкодження передніх відділів спинного мозку проявляється негайно після травми повним паралічем кінцівок і гіпестезією до рівня ураженого сегмента, причому відчуття руху і положення кінцівок і частково вібраційна чутливість збережені. Цей синдром може бути наслідком також згинальній травми. У патогенезі його особливе значення має здавлення передніх відділів спинного мозку зміщеним назад тілом хребця, що посилюється натягом зубовидних зв'язок і деформацією бічних відділів мозку. Якщо при цьому ретельне -рентгенологіческое дослідження виключає пошкодження кістки, то слід запідозрити гостре килоподібну випадання вкінці міжхребцевого диска. Відсутність блоку при ликвородинамических пробах не виключає постійно існуючої передньої компресії спинного мозку, і в цих умовах виникають показання до ламінектомії з перетином зубовидних зв'язок. У подібних випадках іноді доводиться робити пневмоенцефалографія, уточнюючу ступінь і локалізацію зміщення передніх структур пошкодженого хребця і випинання зруйнованих дисків в просвіт хребетного каналу. Поразка передніх відділів спинного мозку при ускладнених пошкодженнях хребта зустрічається часто, і спостерігається, за даними Я. Л. Цив'яна з співавт. (1976), у 4 / s хворих з ушкодженнями хребта і спинного мозку. У подібних випадках, якщо після скелетного витягування і форсованого вправляння протягом доби намітиться хоча б невеликий регрес неврологічної патології, що свідчить про можливість відновлення функції спинного мозку, найбільш доцільною є операція передньої декомпресії спинного мозку, зі стабілізацією передніх структур пошкодженого відділу хребта.
Розлади кровообігу в спинному мозку
В минулі десятиліття патологія спинного мозку при травмі хребта розглядалася в основному як механічне пошкодження. Однак в останні роки висуваються концепції, що підкреслюють значення розлади кровопостачання певних сегментів мозку з розвитком ішемії, тканинної гіпоксії та аноксії з випаданням спінальних. функцій.
Експериментальні, патологоанатомічні та клінічні дані показують, що порушення кровообігу в. спинному мозку може виникнути при струсі спинного мозку і розглядається як рефлекторне. При цьому вазомоторні порушення, стази, диапедезного характеру плазморея з розвитком набряку мозку і петехіальні крововиливи порушують кровопостачання нервової тканини і можуть привести до тканинної гіпоксії, вторинним паренхіматозним некрозів і розм'якшення. Механічні впливу на спинний мозок при зсувах хребця або пролапсе диска поряд з пошкодженням тканини мозку супроводжуються здавленням або розривом кровоносних судин цій галузі і рефлекторними порушеннями кровообігу в сусідніх або віддалених сегментах мозку внаслідок патологічних імпульсів, що виходять із зони пошкодження. При цьому слід врахувати і можливість здавлення розташованої в області пошкодження хребта добре розвиненою корешковой артерії, що має велике значення в кровопостачанні мозку.
Ці концепції підтверджуються клінічними спостереженнями, згідно з якими рівень ураження спинного мозку іноді не відповідає рівню ушкодження хребта.
У ряді випадків рівень сегментарної патології спинного мозку відповідає зазначеному рівню, але при цьому виявляється другий рівень поперечного ураження спинного мозку, розташований значно нижче або вище рівня ушкодження хребта.
Так, наприклад, при пошкодженні шийного відділу хребта і спинного мозку часто виявляються два рівня ураження :
- переважно сегментарного характеру в області верхніх кінцівок,
- поперечне ураження спинного мозку в області ThiV сегмента внаслідок порушення кровообігу мозку на стику постачання двома артеріальними системами.
Найбільш часто спінальна патологія, яка не відповідає рівню ушкодження хребта, виникає на рівні Cv, Thiv, Thxii і Li сегментів, що пояснюється існуванням так званих критичних зон кровообігу на стику двох артеріальних систем спинного мозку, найбільш схильних до декомпенсації при розладах кровообігу.
Порушення гемодинаміки ведуть до ішемічного розм'якшення спинного мозку, частіше за все у випадках «мінімального кровопостачання» в так званих небезпечних, або критичних, зонах.
Анатомічними дослідженнями встановлено, що кровопостачання спинного мозку здійснюється не сегментарной системою корінцевих артерій, а тільки одиничними, добре розвиненими артеріальними стовбурами. Легко виражені порушення кровопостачання викликають лише функціональні явища випадання. Порушення середнього ступеня обумовлюють в першу чергу пошкодження центральних відділів з подальшим розвитком некрозів, розм'якшення і кіст, а важкі ішемії призводять до розладу функцій всього поперечника спинного мозку.
Поразка кінського хвоста і конуса при переломах поперекових і крижових хребців
Ця поразка веде до появи корінцевих симптомів, до розвитку синдрому ураження кінського хвоста або конуса спинного мозку. Слід зазначити, що при відсутності найближчим часом після травми неврологічної симптоматики у віддалені терміни може виникнути корінцевий синдром та клінічна картина міжхребцевого остеохондрозу. Природно, що при переломах хребта може спостерігатися не тільки пошкодження спинного мозку або його корінців, але і поєднане ушкодження сплетінь, симпатичних утворень і нервів кінцівок (особливо при супутніх переломах кінцівок).
Методика обстеження хворого та принципи лікування
Найбільш доцільним при лікуванні ускладнених переломів хребта є спільна робота невропатолога, ортопеда і нейрохірурга. Обстеження хворого має на меті визначення ступеня і характеру ураження нервової системи, деформації хребта, общесоматического стану, виключення супутніх ушкоджень кінцівок і внутрішніх органів.
Клінічна картина переломів характеризується хворобливістю в області пошкодження при пальпації, деформацією (наприклад, утворенням гострого кутового кіфозу — горба при компресійному переломі в грудному відділі), напругою м'язів шиї або спини. У разі зміщення вперед трьох верхніх шийних хребців деформація легко встановлюється пальпацией через рот. При виражених симптомах ураження певного рівня спинного мозку або його корінців топический діагноз пошкодження хребта може бути поставлений з більшою часткою ймовірності з урахуванням неврологічної симптоматики. Рентгенографія хребта проводиться в умовах, що запобігають посилення дислокації хребта.
Лікувальні заходи при переломах хребта зводяться до наступного.
- Транспортування хворого в лікувальний заклад здійснюється таким чином, щоб не посилити деформацію хребта і не викликати вторинних ушкоджень спинного мозку. Найбільш доцільна в разі пошкодження шийного відділу хребта негайна фіксація хворого в рамі стрікер, до якої прикріплено пристосування для скелетного витягування.
- У лікувальному закладі потерпілого з такими ж застереженнями укладають на жорстку постіль або на щит, зверху якого поміщають щільний або повітряний матрац і туго натягнуту (без всяких складок) простирадло. Найбільш доцільно використовувати ліжко зі спеціально повертається двостулкових рамою стрікер. Вона забезпечує хорошу іммобілізацію, витягування, полегшує повертання хворого, зміну білизни і догляд за шкірою ,, спорожнення кишечника, а також перевезення в інше приміщення.
- У лікувальному закладі повинні проводитися ортопедичні заходи, щоб усунути деформацію хребта (особливо просвіту хребетного каналу), забезпечити його стабільність і запобігти вторинне зміщення. Спинний мозок в більшості випадків пошкоджується в момент травми, а подальше здавлення мозку зміщеними хребцями лише посилює це пошкодження.
Природно, що здавлення пошкодженого в момент травми спинного мозку сместившимися частинами хребців, міжхребцевих хрящем, розташованим в межах хребетного каналу, набряклими тканинами, а іноді і гематомою є ускладнює фактором, який погіршує стан спинного мозку і має бути усунутий можливо раніше за допомогою ортопедичних втручань або хірургічним шляхом.
Це досягається наступними лікувальними заходами:
- одномоментним закритим вправлення переломовивихів хребта,
- витяжкою ,
- відкритим (оперативним шляхом) вправлення цих переломовивихів (відкритої репозиції),
- операцією задньої або передньої декомпресії,
- тривалою іммобілізацією хребта, що досягається або оперативним шляхом ( операцією заднього або переднього спондилодезу), або накладенням фіксуючих пов'язок (гіпсових і т. д.).
Хірургічне втручання повинно відповідати таким вимогам:
- повноцінної декомпресії спинного мозку і його судин,
- відновленню нормальних анатомічних співвідношень хребетного каналу і спинного мозку з метою створення оптимальних умов для максимально можливого відновлення функції спинного мозку,
- забезпечення надійної стабілізації пошкодженого хребетного сегмента з метою запобігання вторинним зсувів пошкоджених хребців,
- подальшим функціональним лікуванням для попередження атрофії м'язів, що забезпечують статику хребта під час стояння і ходьби,
- в пізній стадії захворювання, коли вже ясний межа оборотності н євро логічної симптоматики, основне завдання лікаря полягає в створенні умов для максимального використання залишкових функцій, тому тут основними є ортопедичні заходи.
Особливе місце серед пошкоджень хребта займають переломи і вивихи двох верхніх шийних хребців, що обумовлено як особливостями їх топографічних взаємовідносин, так і небезпекою пошкодження довгастого і спинного мозку з летальним результатом.
В атланто-осьовий області зустрічаються:
- травматичний передній вивих або підвивих атланта без перелому зубовидних відростка,
- перелом зубовидних відростка без зміщення,
- переломовивіх атланта і зубовидних відростка,
- перелом атланта.
Дислокація (зміщення) в атланто-аксиальном зчленуванні може бути також наслідком гострих або хронічних інфекційних процесів (переважно ревматичних артритів або запальних процесів в назофарингеальної області), що викликають розслаблення періартикулярних тканин цього зчленування, або ж вроджених аномалій атланта і епістрофея (епіфізеальное поділ зубовидних відростка), відсутність епістрофея, каліцтва атланта.
Лікувальні заходи при переломі і вивихах двох верхніх шийних хребців включають тривале скелетневитягування за звід черепа, а в деяких випадках оперативне втручання з метою ліквідації компресії спинного мозку і забезпечення стабільності в атланто-окціпітальной зчленуванні. В останнє десятиліття увага прикута до так званої гіперекстензіонной травмі шийного відділу хребта (підвидом якої є так звана хлистова травма). Ці ушкодження виникають при транспортних (особливо автомобільних), футбольних травмах, при пірнанні, падінні з висоти, зі сходів вперед особою, при ускладненій інтубації трахеї. При цьому розвивається так званий гострий шийний синдром, виражений в різному ступені і виникає після форсування гиперєкстензии шиї, що перевищує анатомо-функціональні межі рухливості цього відділу хребта. На спонділограммах при цьому часто не вдається виявити кісткової патології хребта, в більш важких випадках, особливо при автодорожніх подіях з разгибательное механізмом насильства, виникають переломи шийних хребців і пошкодження зв'язкового-дискового апарату.
Клінічно ця травма проявляється в різному ступені тяжкості синдромами ураження нервової системи, серед яких розрізняють:
- Корінцевий синдром ( який має місце приблизно в 25% випадків), що виявляється болями в шийно-потиличній області протягом тижнів, а іноді місяців.
- Синдром часткового порушення функцій спинного мозку з наявністю пірамідного синдрому (спостережуваного також приблизно в 25% випадків ). При цьому характерно виникнення пекучих минущих болів в руках внаслідок ураження задніх стовпів і здавлення корінців Суп і Суш з швидко минущим відчуттям слабкості в нижніх кінцівках.
- Синдром поперечного ураження спинного мозку, що виявляються приблизно в 30% випадках. У тих випадках, коли цей синдром є нестійким і швидко регресує, є підстави вважати його проявом спинального шоку. При частковому регресі цього синдрому залишаються стійкі порушення функції спинного мозку різного ступеня вираженості.
- Синдром передньої спінальної артерії виявляється приблизно в 20% випадків і проявляється дистальними парезами верхніх кінцівок з гіпотонією і гіпотрофією м'язів, нижнім парапарезом, дистантних і. диссоційованними розладами чутливості, розладами функції тазових органів.
При гіперекстензіонной травмі спостерігається більш швидке і повне відновлення рухів в нижніх кінцівках (у порівнянні з верхніми) внаслідок переважного ураження передніх рогів шийного потовщення і внутрішніх відділів пірамідного пучка, де розташовуються волокна для верхніх кінцівок. Іноді на тлі швидкого і майже повного регресу вираженого тетрапареза тривалий час все ж відзначається паретичной верхніх кінцівок з атрофією м'язів, особливо дрібних м'язів кисті, фибрилляцией в м'язах плечового пояса і легкої гиперстезии в області передпліч.
Лікування травм хребта і спинного мозку
Лікування хворого, який отримав (навіть приблизно) травму хребта, а також з підозрою на травму спинного мозку, починається в момент його виявлення і ще до доставки його в лікарню. Перше необхідний захід — це іммобілізація хребта по всій довжині. Переважно транспортування травмованого в відділення нейрохірургії або ж багатопрофільне відділення з можливістю лікування спинальних хворих.
У багатьох випадках травми хребта і спинного мозку вимагають хірургічного втручання. Рішення про такий фахівець приймає на підставі ступеня вираженості неврологічних симптомів. Операція, при її необхідності, проводиться в найкоротші терміни, оскільки через 6-8 годин після факту здавлювання спинного мозку і забезпечують його роботу судин результати ішемічних змін можуть бути незворотні. З цієї причини всі присутні на момент госпіталізації пацієнта протипоказання до оперативного втручання усуваються в рамках інтенсивної терапії. Така, як правило, включає в себе оптимізацію роботи дихальної та серцево-судинної системи, показників гомеостазу з точки зору біохімії, усунення (частково або, при можливості, повне) набряку мозку, профілактику інфекцій та ін. Операція може складатися в видаленні, протезуванні або корекції положення (вправленні, декомпресії, реклинацию) хребців, відновлення цілісності пошкоджених органів та інших діях, які забезпечують оптимально можливий зв'язок відділів хребта і спинного мозку.
У разі, якщо травма не вимагає оперативного втручання, лікування полягає в фіксації хребта в природному положенні (з попереднім вправлением, якщо таке необхідно) і стимуляції процесів регенерації тканин, нервових закінчень і функціонування органів , чия робота була порушена внаслідок самої травми або її ускладнень. У комплекс лікувальних заходів нерідко входить і розробка м'язів навколо пошкодженого відділу, теплові процедури і масаж, в більш складних випадках йдеться про іммобілізації хребта в зонах ураження, витягнення. Результат лікування визначає комплекс реабілітаційних заходів.
Протягом останніх півтора десятиліть спостерігається тенденція до переходу від консервативних методів лікування гіперекстензіонной травми шийного відділу хребта (іммобілізація шийно-потиличної області пов'язкою з подальшою фізіотерапією, накладення торакокраніальной пов'язки, при показаннях — витягування) до оперативного втручання у випадках, коли є підстави вважати вплив факторів, що викликають компресію спинного мозку [Іргер І. М., Юмашев Г. С., Румянцев Ю. В., 1979, Schneider et al., 1954, 1971, Schlosbree "1977].
Догляд за хворими з ушкодженнями хребта і спинного мозку дуже важкий для обслуговуючого персоналу, особливо при відсутності регресу важких неврологічних розладів.
Одним з найбільш частих і загрозливих ускладнень при травмі спинного мозку є порушення функції сечового міхура.
Для Терміново спорожнення сечового міхура застосовуються три способи:
- періодична або постійна катетеризація,
- ручне спорожнення сечового міхура,
- прокол міхура.
Для виведення сечі з міхура протягом тривалого часу застосовуються два методи:
- дренування по Монро з використанням припливно-отливного дренажу,
- надлобковая цистостомія.
Дренування по Монро полягає в періодичному надходженні в сечовий міхур слабкого антисептичний розчину або розчинюючої сечові солі рідини, виведенні її з міхура за допомогою системи та «розриву» сифона після спорожнення сечового міхура . Клінічні спостереження показують, що система Монро не попереджає повністю інфікування сечовивідних шляхів, але в порівнянні з іншими методами затримує його розвиток, зменшує його прояви і забезпечує відновлення сечовипускання по так званому автоматичному типу. У тих випадках коли є підстави припускати тривале порушення функції сечовипускання, застосовується метод накладення надлобкового свища.
Основною причиною виникнення і розвитку пролежнів в областях, де внаслідок травми спинного мозку порушена іннервація, є висока чутливість дистрофічних тканин до механічних і інфекційних впливів. Однак в місцях, що не піддаються тиску, ніколи не виникає пролежнів при будь-якої тяжкості ушкодження спинного мозку. При лікуванні пролежнів важливо створити умови, що запобігають труднощі лімфо і кровообігу в уражених тканинах і стимулюючі ці процеси. Для цієї мети застосовують різні мазеві пов'язки (до складу яких іноді входять антибіотики), УФО (ерітемние дози), видалення струпов, висічення некротизованих тканин. При розвитку глибоких пролежнів рекомендується освіження рани, поетапне висічення некротизованих тканин з ранньої або пізньої шкірної пластикою, а при остеомієліті — видалення підлягає кістки.
Реабілітація травм хребта і спинного мозку
З точки зору процесу реабілітації, найбільшу увагу слід приділити пошкоджень хребта, пов'язаних з порушеннями цілісності і функціональності хребців. План реабілітації та комплекси заходів різняться в залежності від стабільності пошкодження. Так, у разі, якщо демонструється тенденція до зміщення хребця (нестабільне ушкодження), реабілітація ґрунтується на фіксуванні такого. Травма, що виражається в клиноподібної компресії, відриві передніх кутів тіла кістки, не вимагає фіксації і може включати більш широкий спектр вправ. Кожен з використовуваних сьогодні методів застосовується строго відповідно до показань і на підставі результатів огляду хворого. При цьому всі підходи спрямовані на зміцнення м'язів тулуба для створення 'м'язового корсету', включають ЛФК, фізіотерапію і механотерапію. При виникненні ускладнень, показана електроімпульсна терапія, стимуляція метаболічних процесів, а також кровообігу і регенерації.
Реабілітація після травм, що призвели до порушення функціонування хребта і спинного мозку , різниться в залежності від ступеня отриманих ушкоджень. У більшості випадків метою реабілітації є максимально повне відновлення частково або повністю втрачених або пригноблених, а також розробка збережених функцій спинного мозку. Найменш оборотні наслідки травми наступають в разі функціонального або анатомічного розриву. В цьому випадку лікувальні та відновлювальні заходи спрямовуються на вироблення функцій, що забезпечують пристосування організму до нових для нього умов. Крім того, завданням фахівців є забезпечення максимально повного зв'язку між відділами спинного мозку.
Усі заходи по реабілітації пацієнтів припускають поступове нарощування навантажень до оптимального рівня. У кожному разі термін закінчення процесу відновлення індивідуальний, проте рідко не перевищує 2-3 місяців. Зокрема, першу половину першого місяця реабілітація має на меті відновлення роботи серцево-судинної і дихальної системи, підняття тонусу пацієнта, профілактику погіршення стану м'язів тіла. Надалі до кінця першого місяця (в залежності від пошкоджень, цей період може збільшуватися) дії персоналу і хворого спрямовані на відновлення роботи інших внутрішніх органів, стимуляцію природної регенерації, підготовку м'язів і всього організму до розширення комплексу рухів.
