
У крові дорослої здорової людини виявлено кілька типів , що розрізняються амінокислотним складом поліпептидних ланцюгів.
Основний гемоглобін дорослої людини — гемоглобін А ( HbA ) (від латинського слова adultus — дорослий) становить 96 — 99% всього гемоглобіну. Його білкова частина складається з двох альфа -ланцюгів і двох бета -ланцюгів.
Гемоглобін Н b А2 містить по дві альфа- і дельта-поліпептидні ланцюги. Його кількість не перевищує 2,5% загального гемоглобіну.
Гемоглобін типу HbF , фетальний (від латинського foetus — плід) складається з двох альфа- і двох гамма-поліпептидних ланцюгів. У крові дорослої людини він міститься в кількості не більше 1,5% від усього гемоглобіну. А в крові плоду він становить основну масу гемоглобіну. Фетальний гемоглобін краще фіксує кисень, що має значення у внутрішньоутробний період розвитку. У новонародженої дитини його зміст досягає 80% від загального гемоглобіну. До кінця першого року життя він майже повністю замінюється на гемоглобін А.
Крім того, в крові людини відкрито значну кількість мутантних гемоглобинов , що утворюються в результаті заміни навіть однієї будь-якої амінокислоти в молекулі гемоглобіну на іншу. Деякі з цих мутацій призводять до важких захворювань — гемоглобінопатії.
Прикладом однієї з різновидів гемоглобинопатии може служити серповидно-клітинна гемолітична анемія. Вона обумовлена появою гемоглобіну типу HbS, в якому в порівнянні з гемоглобіном А в бета-ланцюгах замінюється всього одна амінокислота — глутамінова на валін. При низькому парціальному тиску кисню після віддачі кисню в тканинах, гемоглобін HbS перетворюється в погано розчинну форму і випадає у вигляді веретеноподібних кристалів, які деформують (серповидна форма) і призводять до їх масивного руйнування (гемолізу).
похідні гемоглобіну
Відповідно до функціонального призначення в фізіологічних умовах в організмі утворюються такі його похідні , як: оксигемоглобін , карбгемоглобін , і в невеликій кількості — карбоксигемоглобин та метгемоглобін .
Оксигемоглобін
Оксигемоглобін (Н bO 2) утворюється при збагаченні крові киснем в легенях, за рахунок приєднання кисню до гему гемоглобіну за допомогою координаційних зв'язків заліза без зміни валентності заліза.
Оксигемоглобін дуже лабільний з'єднання і, вступаючи з кров'ю до тканин, де тиск кисню значно нижче, ніж в легенях, він легко дисоціює на вільний гемоглобін і кисень.
карбгемоглобін
Кисень утилізується в процесі клітинного дихання, а гемоглобін вступає в з'єднання з вуглекислим газом, утворюючи карбгемоглобін (Н b З O 2 ) . Це також безсила з'єднання, яке легко віддає вуглекислий газ при надходженні крові в легені. Зв'язок з вуглекислим газом ніяк не гем, а глобин за рахунок вільних аміногруп амінокислот.
Карбоксигемоглобин
Карбоксигемоглобин (Н b СО) — це з'єднання гемоглобіну з окисом вуглецю. Утворюється в результаті дисиміляції гемоглобіну і в нормі не перевищує 2% від загальної маси гемоглобіну.
Карбоксигемоглобин, на відміну від інших похідних гемоглобіну, є дуже міцним з'єднанням. СО утворює ковалентний зв'язок з гемоглобіном , блокуючи асоціацію з ним кисню . Спорідненість СО до гемоглобіну більш ніж в 200 разів вище спорідненості кисню .
При отруєнні чадним газом (СО), коли утворюються значні кількості карбоксигемоглобіну, гемоглобін втрачає здатність зв'язувати кисень.
Крім того, СО здатний потрапляти в клітини і пригнічувати дихальну ланцюг в мітохондріях. Все це призводить до тканинної гіпоксії, ацидозу, депресії центральної нервової системи, блокаді дихального центру і настає смерть від задухи.
Метгемоглобін
Метгемоглобін (Метн b ) -Образ при окисленні двовалентного заліза гема в тривалентне. При цьому наголошується і перехід забарвлення з червоною в коричнево-зелену.
У нормі метгемоглобін утворюється за рахунок процесів аутоокисления і не перевищує 2% від загальної маси гемоглобіну, так як невелика кількість утвореного тривалентного заліза до двовалентного відновлює фермент метгемоглобін-редуктаза. Цей фермент дозволяє підтримувати рівень MetHb в крові на дуже низьких цифрах.
При патології кількість МетНb в крові зростає.
Розвивається стан метгемоглобінемії, яке може бути наслідком генетичної патології — генетично зумовленим дефіцитом MetHb-редуктази, або носити набутий характер. Наприклад, при отруєнні оксидами азоту, ціанідами і ін. Утворені значні кількості метгемоглобіну можуть викликати смерть від нестачі кисню, так як метгемоглобін позбавлений здатності зв'язуватися з киснем.
Хвороба Кастелмана причини, класифікація, симптоми, діагностика, лікування
Хвороба Кастелмана — лімфопроліферативних захворювання невідомої етіології, симптоми, перебіг і прогноз залежать від гістологічного типу і локалізації .
Синоніми: giant lymph node hyperplasia, angiofolicular lymph node hyperplasia.
Хвороба Кастелмана — це
рідкісне захворювання з пошкодженням лімфатичних вузлів у вигляді незлоякісною ангіофолікулярной вузловий гіперплазії.
Іноді хвороба Кастелмана зараховують до гранулематозним захворювань з неясною етіологією (, гістіоцитоз Х, гранулематоз Вегенера).
Доктор Берджамін Кастелман вперше опублікував дані про раніше невідомої патології в 1956 році.
Локалізіровання форма зустрічається у більш молодих пацієнтів у віці близько 40 років, в той час як генералізована — в 50-60 років. На початку не має ні злоякісний, ні моноклональних характер проліферації, але пізніше може в нього трансформуватися.
У США частота 2,1 випадок на 100 тис. Населення.
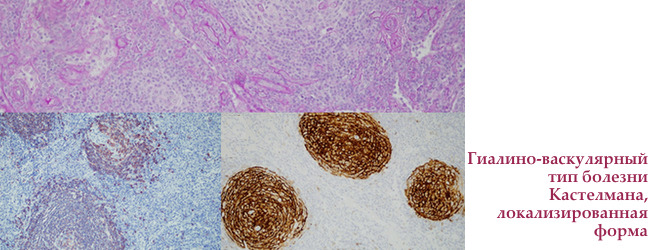
Етіологія хвороби Кастелмана
Причина захворювання неясна. Можлива зв'язок з інфекцією людським герпесвірусом 8 типу (HHV-8), відомого також як вірус герпесу асоційований з саркомою Капоші.
Гістологічні типи хвороби Кастелмана
- гіалін-васкулярної тип — тільки при уніцентріческом формі хвороби
- Плазмоклітинні тип — потенційно агресивний, при локалізованої і генералізованої форми
- змішаний тип
Хвороба Кастелмана за клінічним перебігом
- локалізована (уніцентріческом)
- генералізована (мультіцентріческого)
Унічентріческій гіалін-васкулярної тип хвороби Кастелмана
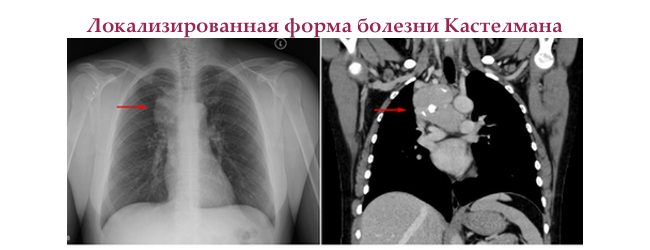
Зустрічається у обох статей, середній вік захворювання 40 років. Пошкоджує один лімфатичний вузол або одну лімфатичну область. Патологічно змінені вузли збільшені до 6-7 см в будь-якій області — від ліктьових ямок і до паху.
Перебіг цієї форми хвороби Кастелмана безсимптомний. Але, при значному збільшенні лімфатичного вузла розташованого поблизу внутрішніх органів розвивається компресійний синдром. У 70% випадків збільшуються лімфатичні вузли в грудній порожнині, що призводить до появи плеврального випоту.
На КТ і МРТ спостерігають посилення, що при лімфомах не зустрічається.
Лікування локальної форми хвороби Кастелмана — повне хірургічне видалення. При субтотальної резекції можливі рецидиви.
Плазмоклітинні тип хвороби Кастелмана
плазмоцитарна варіант хвороби Кастелмана на відміну від описаного вище генералізований, пошкоджує відразу кілька лімфатичних вузлів, частіше поза грудної порожнини. На КТ проявляється у вигляді м'якої тканинної маси з помірним або середнім контрастним підсиленням.
Плазмоклітинні варіант більш агресивний в ставленні до навколишніх тканин, в які може проростати. Найчастіше супроводжується системними симптомами: загальна слабкість, лихоманка, .
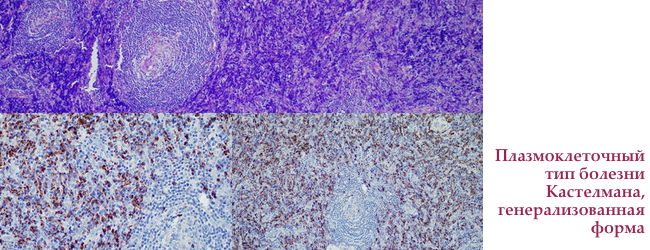
Локалізована плазмоцитарна форма хвороби Кастелмана
уніцентріческом плазмоцитарна форма становить 20% випадків хвороби Кастелмана. Типові В-ознаки — підвищення температури тіла, зниження ваги, нічна пітливість, іноді збільшення печінки або селезінки. У анемія зі зниженням рівня і , тромбоцитопенія (зниження числа ).
мультіцентріческого форма хвороби Кастелмана
мультіцентріческого форма хвороби Кастелмана зустрічається у пацієнтів у віці 50-60 років.
Симптоми:
- збільшення печінки або селезінки
- загальні симптоми — знижена працездатність, лихоманка, постійна втома
- в загальному аналізі крові анемія
- поліклональна гипергаммаглобулинемия
- гранулоцітоз — підвищення числа гранулоцитарних (переважно )в
Перебіг з повторними рецидивами, рідше у вигляді стабільного персистирующего захворювання. Ймовірна трансформація в злоякісну лімфому.
Як і злоякісні лімфоми мультіцентріческого форма хвороби Кастелмана ділиться на стадії. Прогноз несприятливий.
Лікування хвороби Кастелмана
- хірургічне лікування — тільки при локальних формах, при генералізованої формі хвороби Кастелмана — тільки з метою діагностики
- цитостатичні препарати, як і в лікуванні злоякісної неходжкінської лімфоми — CHOP (адриамицин, вінкристин, циклофосфамід, преднизон), CVAD (циклофосфамід, вінкристин, адріаміцін, дексаметазон) з50-67% успішністю
- глюкокортикоїдних гормони — в деяких випадках переводять хвороба Кастелмана в ремісію і зменшує симптоми
- інтерферон альфа
- ALL-трансретинова кислота (ATRA)
- талідомід
- моноклональні антитіла проти інтерлейкіну-6 і рецепторівінтерлейкіну-6
- рітуксімаб
- противірусні препарати
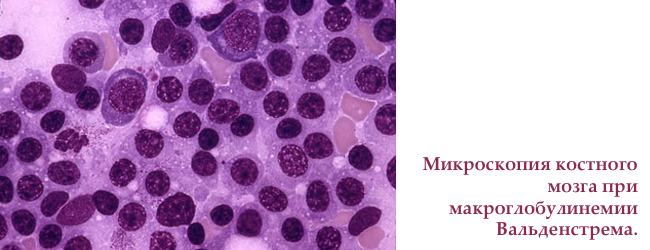
Макроглобулінемія Вальденстрема симптоми, класифікація, діагностика, лікування
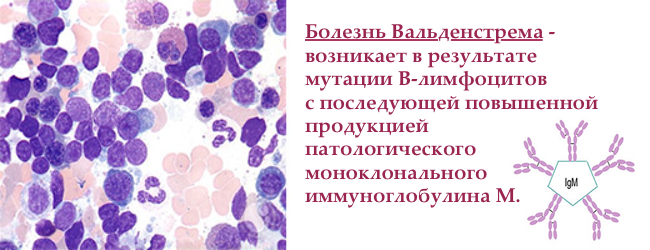
Макроглобулінемія Вальденстрема — рідкісне захворювання , викликане злоякісної мутацією В-лімфоцитів ( ), які виробляють моноклональних макроглобулин М (М-IgM). Гістологічно — лімфоплазмоцитарна лімфома з продукцією М-IgM . Відноситься до групи моноклональних гаммапатій, як і множинна мієлома.
Зустрічається у осіб старшого віку. Основні симптоми хвороби Вальденстрема — порушення зору, неврологічні розлади, носові кровотечі, збільшення печінки і селезінки, лімфатичних вузлів, і підвищена кровоточивість.
У лікуванні використовуються цитостатики, симптоматична терапія, плазмаферез, специфічні анти-CD20 антитіла проти пухлинних клітин.
На 10-11 випадків множинної мієломи припадає 1 випадок хвороби Вальденстрема.
Історія вивчення хвороби Вальденстрема
у 1944 році шведський професор Ян Госта Вальденстрем описав двох пацієнтів з крововиливами в ротовій порожнині, частими носовими кровотечами, , анемією, тромбоцитопенією , і підвищенням і високою в'язкістю плазми крові. В кістковому мозку спостерігалося збільшення числа лімфоїдних клітин. Підвищену густоту крові він пояснив аномально високим рівнем високомолекулярних білків, а відсутність болів в кістках кардинально відрізняло нове захворювання від відомої раніше множинної мієломи.
Епідеміологія хвороби Вальденстрема
Хвороба Вальденстрема зустрічається рідко, 3 нових випадки на 1 млн жителів у рік, що складає 1-2% серед всіх гематологічних новоутворень. Середній вік появи перших симптомів 73 роки, з незначний переважанням жінок (> 3,8, +5,4). У віці до 50 років хвороба Вальденстрема практично не зустрічається, але після 80 років частота підвищується до 28,5 випадків на 1 млн населення. Подібну вікову динаміку можна пояснити поступовим накопиченням помилок в імунній системі при старінні.
Причина хвороби Вальденстрема не відома. Але, наявність В-лімфопроліферативного захворювання у прямого родича (хвороба Вальденстрема, хронічний лімфолейкоз, множинна мієлома, моноклональних гаммапатія неясного значення, злоякісна неходжкінська лімфома) підвищує ризик появи захворювання.
Відноситься до доброякісних, повільно прогресуючим захворюванням, але може мати і тяжкий перебіг.
Симптоми хвороби Вальденстрема
всі симптоми хвороби Вальденстрема можна розділити на три групи :
- токсичність моноклонального імуноглобуліну
- недостатність функції червоного кісткового мозку
- внекостномозговая проліферація лімфоплазмоцітарной лімфоми

Цитокіни,виробляються пухлинними клітинами, призводять до появи таких симптомів:
- цитопения — зниження числа клітин крові
- субфебрилітет і фебрилитет — підвищення температури тіла до 38 і вище градусів
- нічна пітливість
- зниження ваги
- збільшення лімфатичних вузлів
- збільшення печінки і селезінки
Симптоми хвороби Вальденстрема, викликані моноклональних імуноглобуліном М
80% молекул знаходяться всередині судин, асиметричний, великого розміру. Підвищують в'язкість крові в більшій мірі, ніж інші види імуноглобулінів (, ), ушкоджують мікроціркуляціонное русло.
Симптоми високої в'язкості крові — гіпервіскозний синдром — з'являються при щільності плазми в 4 рази перевищує щільність води, що відповідає концентрації М-IgМ більше 30 г / л.
Прояви гіпервіскозного синдрому
- спонтанні кровотечі з носа і ясен
- постійний головний біль в результаті підвищеного внутрішньочерепного тиску
- зниження зору і слуху
- запаморочення
- зниження пам'яті, інтелекту аж до недоумства
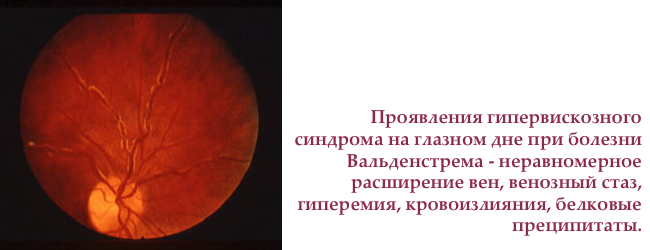
- тромбоз центральної вени сітківки
Сітківка ока — єдине місце в тілі людини, де можна безпосередньо оглянути капіляри. Огляд очного дна — простий і доступний метод дослідження пошкодження судин викликаного гіпервіскозним синдромом.
- задишка
Наявність симптомів при хворобі Вальденстрема не залежить безпосередньо від в'язкості крові.
Кріоглобулінемія
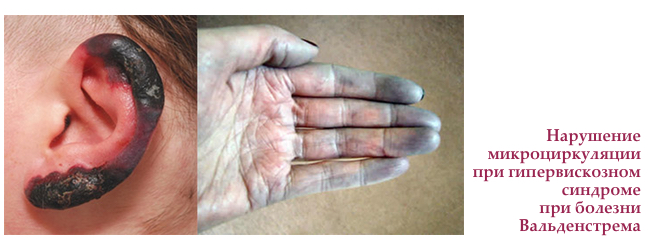
Кріоглобулінемія — стан, при якому білки крові випадають в осад (кріопреціпітація) всередині судин при зниженні температури тіла нижче 37 ° С. Кріоглобулінемія при хворобі Вальденстрема розвивається в 10-20% випадків і проявляється у вигляді збліднення або посиніння пальців рук і ніг, кінчиків вух або носа після переохолодження.
макроглобулин може взаємодіяти з деякими чинниками і прикріплятися до поверхні , що призводить до підвищеної кровоточивості.
Відкладення імуноглобуліну М в нирках призводять до , в кишечнику — до . Кожен п'ятий з хворобою Вальденстрема має периферичну демієлінізуючих сенсорну полінейропатію, що супроводжується слабкістю в руках і ногах, тремором, порушенням ходи.
Симптоми хвороби Вальденстрема, викликані інфільтрацією кісткового мозку
- пригнічення нормального кровотворення
- анемія — зниження рівня і / або
- зниження числа тромбоцитів — тромбоцитопенія
- рідше знижується рівень — лейкопенія
Інші симптоми хвороби Вальденстрема
- збільшення лімфатичних вузлів, селезінки, печінки
- хвороба холодових аглютинінів з
- остеолиз скелета без болю в кістках
- інфільтрація інших органів — шкіри, центральної нервової системи, легенів
- легка ланцюг імуноглобуліну Мвідкладається в тканинах і з нього формується (в серці, нирках, печінці, шлунково-кишковому тракті)
- хронічна втома, слабкість і знижена працездатність
- свербіж шкіри
- зниження імунорезистентність — часті інфекційні захворювання, особливо верхніх дихальних шляхів, кишечника, сечівника
Діагностика Вальденстрема
- — зниження рівня гемоглобіну і / або еритроцитів, зниження числа тромбоцитів
- підвищення ШОЕ до 100 мм / год з наступним визначенням імуноглобулінів (кількісно )
- підвищення
- підвищення
- — одноріднийМ-пік між бета і гамма фракцією гаммаглобулінів — це моноклональних імуноглобулін М — не залежно від концентрації служить діагностичним критерієм діагнозу
- , парапротеїнурії
- підвищення густоти плазми крові
- цитогенетическое дослідження — делеция 6q в 50% випадків, прогностичне значення невідомо

- негативний результат дослідженнямутації IgH, t (14,18), t (11,14) — підтверджують діагноз хвороби Вальденстрема
- з мієлограма — інфільтрація кісткового мозку малими лімфоцитами з -плазмоцитоїдні / плазмоцелюлярной диференціації, інтратрабекулярний тип інфільтрації
не проводиться для виявлення хвороби Вальденстрема в зв'язку з низькою інформативністю
- — CD5- +, CD10-, CD19 +, CD20 +, CD22 +, CD23-, CD25 +, CD27 +, FMС7 +, CD103-, CD138 —
- ПЛР-аналіз на точкову мутацію в гені MYD88 — 100% випадків хвороби Вальденстрема
- візуалізація збільшених лімфатичних вузлів, селезінки, печінки — УЗД
Додаткові дослідження при хворобі Вальденстрема
- — , , , ,
- — , ,
- КТ або ПЕТ
- електроміографія
- огляд очного дна
Лікування хворобиВальденстрема
Підтверджений діагноз хвороби Вальденстрема не служив показанням до початку лікування.
Критерії початку лікування хвороби Вальденстрема
- ознаки підвищеної в'язкості плазми крові — зміни на очному дні, неврологічні симптоми
- периферична нейропатія
- амілоїдоз з легких ланцюгів
- симптоматична кріоглобулінемія
- зниження гемоглобіну нижче 100 г / л, тромбоцитів менше100 * 10 9 / л
- значне збільшення лімфатичних вузлів, печінки або селезінки, ознаки трансформації в гігантоклітинний лімфому
- інші симптоми, що торкаються якість життя пацієнта
Лікування хвороби Вальденстрема, як і інших гематологічних пухлин завжди індивідуально і залежить від віку, провідних симптомів, можливостей і досвіду лікувального закладу.
Препарати для лікування макроглобулінемії Вальденстрема
- алкилирующие цитостатики — хлорамбуцил
- глюкокортикоїди — преднізолон, дексаметазон
- аналоги пуринів — флударабин, 2-хлордеоксіаденозін, пентостатином
- рітуксімаб
- талідомід і ревлімід
- бортезоміб
- силденафіл
- іматиніб
При масивної спленомегалії проводиться . При неуспішності лікування макроглобулінемії Вальденстрема або резистентності клону до застосовуваних препаратів рекомендована високодозова хіміотерапія з трансплантацією кісткового мозку .
Плазмоферез використовують для лікування гіпервіскозного синдрому. Обмінюють 1-1,5 обсягів плазми, що знижує в'язкість на 60-75%, ефект настає швидко і триває 4-6 тижнів. Плазмаферез можна проводити довічно.
Несприятливі прогностичні фактори при хворобі Вальденстрема
- вік старше 65 років
- гепато- і спленомегалія
- підвищення бета-2-мікроглобуліну
- анемія — гемоглобін нижче 100 г / л, тромбоцити нижче100 * 10 9 / л
- менше 40 г / л
- рівень імуноглобуліну М не має прогностичного значення

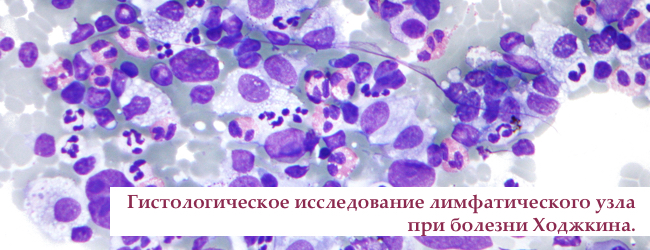
Лімфома Ходжкіна причини і симптоми, класифікація, стадії і аналізи, лікування
Лімфома Ходжкіна — злоякісне захворювання лімфатичної системи, проявляється і загальними симптомами. Основа захворювання — злоякісні клітини Рід-Штернберга і Ходжкіна, виникають з постгермінальних В-лімфоцитів які уникли апоптозу.
На відміну від інших пухлин, III-IV стадії хвороби Хожкіна успішно виліковують.
Синоніми: хвороба Хождкіна, лімфогранулематоз, лимфогранулема, злоякісна гранульома.
Історія
Термін "хвороба Ходжкіна" з'явився в 19-м столітті на честь англійського лікаря, сірка Томаса Ходжкіна, який вперше в 1832 році опублікував роботу про невідомому раніше захворюванні лімфатичних вузлів.
Довгий час лімфома Ходжкіна залишалася невиліковним смертельним захворюванням.
На рубежі 60-70 років минулого століття в протоколи лікування ввели хіміотерапію, що значно поліпшило прогноз хворих. З'явилися в той час лікувальні режими використовуються і сьогодні в різних модифікаціях.
Лімфатична система
Лімфатична система складається з тимуса (вилочкової залози), селезінки, червоного кісткового мозку, л імфатіческіх вузлів і лімфатичних судин . Її основна функція — захист організму від усього чужого — вірусів, бактерій, злоякісних клітин. Тому вона не сконцентрована в одному місці, а "розкидана" по всьому тілу.
Лімфатичні вузли — це невеликі гороховидний освіти, розташовані уздовж лімфатичних судин. Вони перекачують лімфу від периферії тіла до центру, в них дозрівають лімфоцити і проходять більш "вузьку спеціалізацію" на Т- та В- підвиди. Також в них відбувається імунологічна фільтрація для захисту від генетично невідомих речовин (бактерій, вірусів, токсинів).
Лімфома Ходжкіна vs Ні-Ходжкінская лімфома
Обидва види пухлин "виходять" з клітин лімфатичної системи, але для хвороби Ходжкіна характерні зміни саме в В-лімфоцитах, а в групу неходжкінських лімфом включили всі залишилися новоутворення, що не підпали під критерії хвороби Ходжкіна.
Поширеність
Лімфома Ходжкіна зустрічається досить рідко , кожне 100-200 новоутворення припадає на дане захворювання. Виділяють два піки захворюваності — 20-30 років і старше 60 років, з незначним переважанням серед чоловіків (3>: 2+). Частота нових випадків 2,9 на 10 тис. Населення в рік. Одне з найпоширеніших захворювань у молодому віці (19-35 років).
Причини лімфоми Ходжкіна
Точна причина появи лімфоми Ходжкіна не відома. Загальноприйнята теорія двоетапного впливу .
У першій фазі лімфатична клітина на одному з етапів свого розвитку уникне запланованої клітинної смерті (апоптозу) і залишається безсмертної в лімфатичному вузлі. 50% випадків такої метаморфози викликані , але причина решти 50% так і залишається невідомою. Тривалий термін життя даної клітини збільшує ймовірність другого вторгнення в генетичну інформацію, яка і визначить появу лімфоми Ходжкіна.
Субстрат лімфоми Ходжкіна — великі 2-3 ядерні клітини Рід-Штернберга (Березовського-Ріда-Штернберга) і клітини Ходжкіна.
озлокачествлению клітини виробляють велику кількість цитокінів — речовин, місцево змінюють імунну реакцію. Так, навколо клітин Рід-Штернберга з'являється запальний інфільтрат, що дозволяє новоствореним клітинам уникати смерті, ділиться і проникати в інші лімфатичні вузли. Співвідношення між "поганими" і "хорошими" клітинами 1: 100, що не завжди дозволяє їх побачити при мікроскопічному дослідженні. Дуже часто перший результат гістології лимфоузла вкаже тільки на запалення, тому перевіряти потрібно кілька зрізів з декількох лімфатичних вузлів. Мета — знайти клітини клітини Ходжкіна і Ріда-Штернберга, щоб підтвердити діагноз.
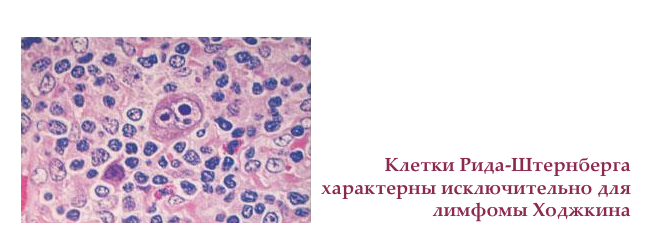
Гістологічна класифікація лімфоми Ходжкіна
- з нодулярним склерозом
- смешанноклеточний варіант
- лімфома Ходжкіна багата лімфоцитами (лімфогістіоцитарні варіант) — немає типових клітин Рід-Штернберга, злоякісні клітини представлені великими клітинами з розділеними лобулярноїядрами
- з придушенням лімфоїдної тканини (лимфоцитарная деплеція), часто в асоціації з ВІЛ-інфекцією
Кожному типу властива певна частота, прогноз, чутливість до лікування.
Симптоми лімфоми Ходжкіна
збільшення лімфовузлів
- безболісне збільшення лімфатичного вузла на шиї, під пахвами або в паху, рідше всередині грудної або черевної порожнини у 7 з 10 пацієнтів
- 90% випадків лімфоми Ходжкіна починається з збільшення лімфовузлів над діафрагмою
- лімфатичний вузол безболісний, тугий як щільна гума, рухливий, "як картопля в мішку"
- збільшений лімфатичний вузол більше 4 тижнів — показання для гістологічного дослідження
В-симптоми
- підвищення температури тіла більше 38 ° C без явних для того причин, постійне і тривале або періодичне
- лихоманкаПеля-Ебштейна — чергування періодів підвищеної і нормальної температури тіла з інтервалами по 3-10 днів
- нічна пітливість з необхідністю міняти постільну білизну
- зниження ваги
- загальна слабкість
Інші симптоми лімфоми Ходжкіна
- здавлення збільшеним лімфовузлом сусідніх органів — кашель і задишка
- свербіжшкіри
- хворобливість збільшених лімфатичних вузлів після алкогольних напоїв — вкрай рідко (але, запоминаемо)
Якщо описані вище симптоми тривають у Вас більше 4-х тижнів — зверніться до лікаря.

Стейджінг лімфоми Ходжкіна
Для визначення стадії захворювання потрібно визначити:
- де в тілі збільшені лімфатичні вузли
- з одного боку діафрагми або з обех сторін (діафрагма — плоска м'яз, що відокремлює грудну порожнину від черевної)
- пошкоджені чи інші органи — печінку, легені,кишечник, кістки і т.д.
Стадії лімфоми Ходжкіна
Поділ на стадії проводиться класифікації Енн-Арбор.
1 стадія — залучена одна група лімфатичних вузлів (I) або один орган за межами ліфмоузлов
2 стадія — вражені 2 більше груп лімфатичних вузлів (II), але тільки з одного боку діафрагми або орган + лімфовузли по одну сторону діафрагми (IIE)
3 стадія — змінені лімфатичні вузли по обидва боки діафрагми, з або без ураження інших органів (IIIE), селезінки (IIIS)
4 стадія — захворювання поширюється крім лімфовузлів ще й навнутрішні органи — печінку, нирки, кишечник, червоний кістковий мозок
У діагнозі лімфоми Ходжкіна також враховують:
- E — поширення пухлинної тканини з лімфатичного вузла на прилеглі органи
- S (spleen) — ураження селезінки
- X — об'ємне утворення великого розміру
Кожну стадію хвороби Ходжкіна ділять на категорії:
- А — відсутність симптомів, тобто лімфома виявлена в найбільш ранній і сприятливою для лікування стадії
- В — присутність одного або декількох В-ознак: чи не цілеспрямоване зниження ваги на більш ніж 10% за останні 6 місяців, фебрильна температура (вище 38 ° C), підвищена пітливість, особливо в нічні години
Діагностика Хождкіна
Мета всіх діагностичних заходів при хворобі Ходжкіна не тільки підтвердити діагноз, а й визначити стадію хвороби і прогноз.
- в першу чергу проводять гістологічне дослідження збільшеного лімфатичного вузла при місцевому знеболюванні
- — в початкових стадіях лімфоми Ходжкіна залишається без змін, наступні зміни не є специфічними
- знижується число лімфоцитів і ростуть в
- принаявності свербіння шкіри — підвищення числа
- (зниження рівня і / або )
- з підвищенням числа і позитивної
- зниження чиста тромбоцитів
- — , ,, , — при їх підвищенні ймовірно ураження печінки
- — , ,
- CОЕ підвищено
- підвищено
- — до 50г / л
- — підвищена
- — CD30 +, CD15 +, CD20-
- рентгенологічне дослідження органів грудної порожнини — дозволяє "побачити" збільшені лімфовузли в средостеріі
- УЗДорганів черевної порожнини — стан печінки, розміри селезінки, нирок, підшлункової залози, збільшення внутрішньочеревних лімфатичних вузлів і здавлення ними сусідніх органів
- УЗД лімфатичних вузлів шиї , пахвовій і паховій областей
- комп'ютерна томографія — всього тіла, від шиї до малого таза
- ПЕТ — позитронно-емісійна томографія — краще візуалізує лімфатичні вузли при хворобі Ходжкіна, але не здатне відрізнити запалені лімфатичні вузли від пухлинних
- з мієлограма — гістологічне дослідження стовпчика червоного кісткового мозку отриманого з клубової кістки для виключення захворювання безпосередньо кісткового мозку або проростання лімфоми в кістковий мозок
Додаткові обстеження перед початком лікування
- ЕКГ і ехокардіографія серця
- спирометрия — оцінка функції лікування
- дослідження статевих залоз — аналізи на гормони,
Несприятливі фактори ризику хвороби Ходжкіна
- поразку більше 3-х груп лімфатичних вузлів
- висока
- велика пухлинна маса в середостінні
- поразкуорганів поза лімфатичної системи (буква Е в Стейджінг)
Лікування хвороби Ходжкіна
Лікування хвороби Ходжкіна полягає в комбінації протипухлинної хіміотерапії і променевої терапії. Тип і інтенсивність лікування (число циклів хіміотерапії, доза опромінення) залежать від стадії захворювання, наявності факторів ризику, загального стану і віку пацієнта.
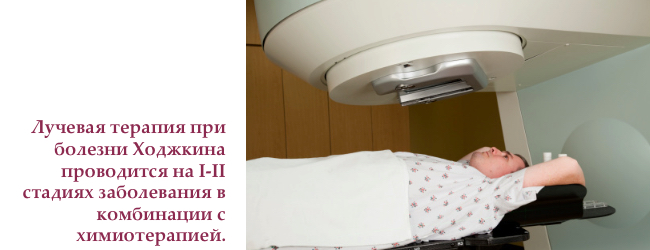
Променева терапія хвороби Ходжкіна
Радіотерапія використовує в високоенергетичне опромінення, що знищує злоякісні клітини і зупиняє їх зростання.
Хіміотерапія
Протипухлинна хіміотерапія хвороби Хождкіна полягає в прийомі лікарських препаратів руйнують клітини пухлини в тілі.
У лікуванні хвороби Ходжкіна завжди використовують комбінації декількох препаратів, які підсилюють дію один одного. Деякі препарати приймаються в формі таблеток, інші вводяться внутрішньовенно. Хіміотерапія проводиться циклами (серіями), коли один і той же перелік препаратів приймають через певні проміжки часу — 2, 3, 4 тижні.

Стандартні режими хіміотерапії хвороби Ходжкіна
- ABVD — адриамицин, блеоміцин, вінбластин, дакарбазін
амбулаторний, без необхідності госпіталізації, добре переноситься пацієнтами, повторюють кожні 14 днів, всі препарати вводять внутрішньовенно протягом одного дня, додатково застосовують ліки проти нудоти і лихоманки
перші три дні лікування медикаменти вводять внутрішньовенно при госпіталізації в лікарню, подальше лікування амбулаторно і полягає в прийомі таблеток 2 тижні, наступний цикл повторюють через 3 тижні
Лікування різних стадій хвороби Хождкіна
- I, II стадії хворобиХоджкіна
- зі сприятливим прогнозом (без факторів ризику) — 2? ABVD + радіотерапія 20 Gy (Грей )
- з несприятливим прогнозом — 2? BEACOPP + 2? ABVD + променева терапія на уражені зони 20 Gy.
- III, IV стадії хвороби Ходжкіна
- 6? BEACOPP в інтенсифікованому режимі
- рецидив хвороби Ходжкіна — охоронний режим (заснований на похідних платини), аутотрансплантация кісткового мозку
Прогноз хвороби Ходжкіна
При локалізованої формі хвороби Ходжкіна успішність лікування 80-90%. Поширену форму (III-IV стадії) інтенсифікований лікувальний режим може вилікувати у 80% пацієнтів, що застосовуються в онкології величезний успіх.
Найбільший ризик рецидиву — повернення хвороби Ходжкіна — в перші 3 роки після закінчення лікування, потім ймовірність наближається до нуля.

Трансплантація кісткового мозку при хворобі Ходжкіна
при безуспішності лікування хвороби Ходжкіна, стійкості до препаратів, повторних рецидивах з необхідністю підвищення доз хіміотерапії проводиться пересадка кісткового мозку.
Побічні ефекти лікування хвороби Ходжкіна
Речовини, що використовуються в лікуванні хвороби Ходжкіна, високоефективні, але їх застосування супроводжується небажаними побічними ефектами.
Побічні ефекти променевої терапії
- втома — найбільш часто, після закінчення лікування проходить, але може тривати до 6 тижнів після закінчення
- відсутність апетиту
- раціаціонний опік шкіри — місця над лімфатичними вузлами, на яке діяли радіаційні промені, від легкого почервоніння до опіку 2 ступеня
- мукозит — запалення слизової оболонкирота, стравоходу при опроміненні області шиї, що проявиться болем при ковтанні, в ротовій порожнині з'являються білястий наліт або почервоніння
- нудота і блювота — меншої інтенсивності, ніж при хіміотерапії
- випадання волосся (алопеція)

Побічні ефекти хіміотерапії хвороби Ходжкіна
- залежать від виду лікування
- нудота і блювота — для зниження даного побічногоефекту хіміотерапії хвороби Ходжкіна профілактично використовують антиеметики — препарати знижують почуття нудоти і бажання вирвати, не рекомендоване їсти дуже теплу або холодну їжу, сильно солодку або пряну
- алопеція
- "втрата" вен на руках після хіміотерапії, що вирішується введенням канюлі в велику вену (на шиї, під ключицею або в паху) або порту для внутрішньовенних інфузій
- зниження фертильності, в деяких випадках безпліддя
Після успішного закінчення лікування хвороби Ходжкіна повторні обстеження у лікаря з комплексом аналізів повинні проводиться,
- 1 рік — кожні 3 місяці
- 2-3 рік — кожне півріччя
- після 3-х років — один раз на рік
Мієлодиспластичний синдром причини, класифікація, симптоми, діагностика, лікування
Мієлодиспластичний синдром — група набутих захворювань кісткового мозку, виникають в результаті мутації стовбурової клітини крові з порушенням її диференціації, що веде до неефективного кровотворенню з ризиком переходу в гострий лейкоз.
Синоніми: МДС, олігобластіческій лейкоз, прелейкоз, myelodysplastic syndrome, MDS.
Міелос — відноситься до кістковому мозку, дисплазія — неправильний розвиток органів і тканин.
Особливості мієлодиспластичного синдрому
- кілька захворювань, об'єднаних в одне для зручності діагностики
- клональное порушення кровотворення з численними диспластическими змінами в кістковому мозку
- хронічний характер, з'являється поволі, а не гостро і раптово
- ймовірність трансформації в більш злоякісний лейкоз
- невідповідність між багатим клітинами (гіперцелюллярний) кістковим мозком (хоча може бути і нормо-і гіпоцеллюлярная) інедоліком клітин крові на периферії
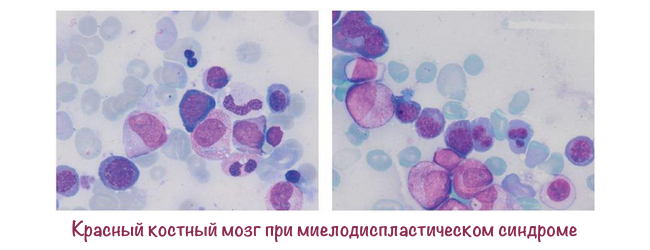
Що таке червоний кістковий мозок?
Червоний кістковий мозок — це м'яка тканина, в якій зі стовбурної клітини утворюються:
- еритроцити — доставляють тканинам кисень, а легким — вуглекислий газ (СО 2 )
- лейкоцити — захищають від інфекцій, кілька підвидів клітин — нейтрофіли, еозинофіли, базофіли, лімфоцити і моноцити
- тромбоцити — кров'яні пластинки, допомагають зупинити кровотечу
в кістковому мозку все клітини проходять кілька стадій розвитку, поступово перетворюючись в ту, яка відправиться «працювати» в русло крові.
Здоровий кістковий мозок містить незрілі клітини — бласти, які потім дозріють в нормальні еритроцити, лейкоцити або тромбоцити. У периферичної крові бластів немає.
Епідеміологія мієлодиспластичного синдрому
Мієлодиспластичний синдром до 40 років зустрічається вкрай рідко, але з підвищенням віку його частота швидко підвищується. Якщо в групі 60-69 років чекають 7 нових випадків на 100 тис. Населення в рік, то після 70 років — до 20-40 / на 100 тис. / Рік.
Етіологія мієлодиспластичного синдрому
Як і у більшості захворювань крові, причина мієлодиспластичного синдрому не відома. Такий МДС називають первинним .
Передбачається несколькоступенчатое дію мутагенного фактора на стовбурові клітини крові для появи порушень в її ДНК. Кістковий мозок починає виробляти структурно і функціонально аномальні клітини, які поступово витісняють нормальні.
Про вторинному мієлодиспластичний синдром говорять, коли відомий фактор, що ушкоджує — лікування цитостатичними препаратами, опромінення, повторний контакт з хімічними речовинами (органічні розчинники, бензен, пестициди).
У МДС була доведена роль інфекції (як наприклад, при лімфомі) або спадковості.
Симптоми мієлодиспластичного синдрому
Симптоми у миелодиспластического синдром дуже різноманітні, що визначено основними змінами в кістковому мозку. У частини пацієнтів може бути тільки незначна анемія, а захворювання тривалий час стабільно і не вимагає ніякого лікування. З іншого боку є і прогностично несприятливі варіанти перебігу хвороби, при яких підвищений ризик переходу МДС в більш важке захворювання — гострий лейкоз.
Якщо в загальному аналізі крові знижена кількість клітин двох ліній (наприклад, еритроцити і тромбоцити) — це біцітопенія , якщо трьох — панцитопенія .
Анемія
Анемія при мієлодиспластичний синдром зустрічається дуже часто, супроводжується зниженням числа еритроцитів і / або гемоглобіну в Загалом аналізі крові. У 80% пацієнтів гемоглобін нижче 100 г / л.
Симптоми анемії
- постійна втома
- знижена працездатність і швидка стомлюваність
- головний біль і шум у вухах
- блідість шкіри і слизових оболонок
- неможливість зосередитися
- задишка при фізичному навантаженні
- прискорене серцебиття
На вираженість симптомів анемії впливають вік пацієнта, швидкість розвитку анемії, супутні захворювання.
Лейкопенія
Зниження числа лейкоцитів в крові — лейкопенія спостерігається в кожному третьому випадку мієлодиспластичного синдрому. Нейтропения — зниження абсолютного або відносного (в лейкоцитарній формулі) числа підвиду лейкоцитів — нейтрофілів.

Нейтрофіли — основні представники лейкоцитів в крові і «займають» від 40 до 74% всієї лейкоцитарної формули. Головне завдання нейтрофілів — захистити організм від інфекцій, переважно бактеріальних і грибкових. Якщо нейтрофілів мало — розвивається імунодефіцит.
Симптоми зниження числа лейкоцитів і нейтрофілів
- частіші бактеріальні або вірусні інфекції
- в оспаленіе легких і бронхіти — підвищення температури тіла, кашель, задишка
- фарингіти, ларингіти, синусити — кашель, біль у горлі, нежить, біль при ковтанні
- запалення середнього вуха — біль у вусі, головний біль,лихоманка
- інфекція сечовивідних шляхів — часте і хворобливе сечовипускання
- інфекції травної системи — пронос, часті випорожнення, метеоризм
- запальні явища на слизовій оболонці рота — гінгівіт, стоматит
- підвищена пітливість
- підвищення температури тіла
- слабкість, втрата ваги
- збільшеннялімфовузлів
При мієлодиспластичний синдром інфекції мають більш затяжний і млявий перебіг, гірше піддаються лікуванню, можуть бути викликані незвичним збудником.
Тромбоцитопения
Тромбоцитопения — зниження числа і порушення функції тромбоцитів. У 10% пацієнтів з мієлодиспластичним синдромом.

Симптоми тромбоцитопенії
- спонтанні кровотечі з носа
- кровотечі з ясен під час чищення зубів
- більш тривалий кровотеча після дрібних хірургічних втручань — видалення зуба, рідної плями
- петехии — дрібні точкові крововиливи на шкірі ніг
- сильніші менструації у жінок
- схильність до появи синців
- рідше — крововилив в головний мозок
Діагностикамієлодиспластичного синдрому
Мієлодиспластичний синдром можна діагностітьвать в фазі змін в загальному аналізі крові, при повній відсутності будь-яких сімпмтомов, а також на стадії ускладнень у вигляді важких інфекцій або кровотеч. Симптомів, які дозволили б запідозрити виключно мієлодиспластичний синдром — немає. Основний метод діагностики — лабораторні дослідження.
- загальний аналіз крові з лейкоцитарною формулою — простий і дуже доступний метод дослідження, що дозволяє запідозрити наявність мієлодиспластичного синдрому
- зниження гемоглобіну і / або еритроцитів — з нормальним або підвищеним MCV (дуже рідко знижений), високим RDW, що свідчить про анизоцитоз — різні розміри еритроцитів
- зниження лейкоцитів, особливо нейтрофілів з появою мієлобластів і більш незрілих елементів, при оцінці лейкоцитарної формули підмікроскопом — аномалії будови ядра, вакуолізація і грануляція
- зниження тромбоцитів
- стернальная пункція або трепанобиопсия — проводяться для більш глибокої верифікації підвиду мієлодиспластичного синдрому, лікар під мікроскопом переглядає як дозрівають клітини в кістковому мозку, оцінює їх число і будова , одночасно проводиться імунофенотіпізація (визначають антигени), цитогенетическое (хромосомні аномалії), цитохимические дослідження, флуоресцентна гібридизація in situ(FISH)
Для оцінки стану інших органів і розрахунку прогностичного індексу при мієлодиспластичний синдром додатково проводять:
- печінкові проби — загальний білірубін, АСТ, АЛТ, ГГТ, лужна фосфатаза
- ниркові проби — креатин, креатинін, сечова кислота
- гормони щитовидної залози
- ферритин — для оцінки обсягу депонованого заліза
- еритропоетин
- ретикулоцити — диференційний діагноз гемолітичної анемії
- вітамін В12 і фолієва кислота — примакроцитоз в загальному аналізі крові
- антитіла до HIV
- аналізи на вірусні гепатити
- згортання крові
- проба Кумбса

Класифікація мієлодиспластичного синдрому
Класифікація мієлодиспластичного синдрому запропонована Всесвітньою організацією охорони здоров'я в 2008 році:
- рефрактерная анемія (РА)
- рефрактерная цитопения з мультилинейной дисплазією (РЦМД)
- мієлодиспластичний синдром зізольованою делецией 5 хромосоми (del 5q)
- мієлодиспластичний синдром некласифікований (МДС-Н)
- рефрактерная анемія з кільцеподібними сидеробластами (РАКС)
- рефрактерная цитопения з мультилинейной дисплазією і кільцевими сидеробластами (РЦМД-КС)
- рефрактерная анемія з надлишком бластів-1 (РАІБ-1)
- рефрактерная анемія з надлишком бластів-2 (РАІБ-2)
У діагнозі часто пишуть не повна назва, а тільки скорочення.
Міжнародна шкала оцінки прогнозу (International Scoring Prognostic System) IPSS
|
Кількість бал ів |
|||||
| Прогностичний фактор | 0 | 0,5 | 1,0 | 1,5 | 2,0 |
| бластів в кістковому мозку (%) |
менше 5 |
5-10 | — | 11-20 |
21-30 |
| Каріотип | сприятливий | проміжний | несприятливий | — |
— |
| Цитопения | 0/1 | 2/3 | — | — |
— |
- низький ризик — 0 балів
- проміжний 1 — 0,5-1,0 балів
- проміжний 2 — 1,5-2 бали, ризик бластоза 10-20%
- високий ризик — більше 2, 5 балів, термін переходу в гострий лейкоз 2 місяці
Рефрактарная анемія (РА) з унілінеарной дисплазією
Характерна стійка до лікування препаратами заліза, фолієвої кислоти і вітаміном В12. Зустрічається найчастіше, з мінімальним ризиком подальшої прогресії. Лікування підтримує — вітаміни, еритропоетин, переливання крові. При симптомах перевантаження залізом — комплексосвязивающіе препарати.
Рефрактерная анемія з кільцеподібними сидеробластами (РАКС)
РАКС входить в групу нізкоагрессівних МДС, а назва походить від типової знахідки в кістковому мозку — сидеробласти — підвид червоних клітин крові містять гранули заліза навколо ядра. У крові анемія, але бластів немає.
Рефрактерная цитопения з мультилинейной дисплазією (РЦМД)
В загальному аналізі крові знижена кількість кількох ліній клітин крові — нейтропенія, тромбоцитопенія, анемія. В кістковому мозку менше 5% бластів і менше 15% пертсневідних сидеробластов. Якщо у пацієнта з РМЦД більше 15% сидеробластов, то говорять про РМЦД-КС (тобто з кільцевими сидеробластами). Лікування залежить від конкретного результату дослідження крові — при анемії лікуємо анемію, при нейтропенії — нейтропенія.
Рефрактерная анемія з надлишком бластів
- РАІБ-1
- РАІБ-2
Розподіл засноване на числі бластів в кістковому мозку: РАІБ-1 — 5-9% бластів, РАІБ-2 — 10-19% бластів у кістковому мозку, палички Ауера. Тяжкість і прогресування даних форм мієлодиспластичного синдрому залежить від числа змін в хромосомах.
Мієлодиспластичний синдром з ізольованою делецией 5q (5q-мінус синдром)
Назва підтипу пов'язано з відсутністю плеча q на 5-й хромосомі. Делеция може бути єдиною хромосомної аномалією або поєднуватися з іншими змінами. В загальному аналізі крові анемія, а число тромбоцитів може бути підвищено. Характерний для осіб середнього і старшого віку. Лікування — леналідомід.
Мієлодиспластичний синдром некласифікований
Ця група потрапляють всі варіанти МДС не потрапили під критерії інших груп. Наприклад, в загальному аналізі крові панцитопенія, але в кістковому мозку недостатньо диспластичность клітин (менше 10%) або зміни тільки в одній лінії клітин. Також сюди віднесені випадки з цитогенетичними аномаліями, типовими для МДС, але без диспластичних змін в кістковому мозку.
Хронічний мієломоноцитарний лейкоз — ХММЛ
Хронічний мієломоноцитарний лейкоз на сьогоднішній день віднесений в мієлодиспластичні / мієлопроліферативні захворювання. При ХММЛ-1 менше 10% бластів у кістковому мозку і більше 1 * 10 9 / л моноцитів в крові. Якщо число бластів досягне 10-20% — це ХММЛ-2.
Прогностична система Всесвітньої організації охорони здоров'я (WPSS)
| Бали | 0 | 1 | 2 | 3 |
| Вид мієлодиспластичного синдрому за класифікацією ВООЗ (читайте вище) | РА, РАКС, 5q — | РАІБ-1 | РАІБ-2 | |
| Каріотип | хороший | середній | поганий | — |
| Потреба в переливанні крові | немає | регулярна | — | — |
Каріотип:
- сприятливий: нормальний, -Y, del 5q, del 20q
- несприятливий: більше 3х аномалій або аномалії 7 хромосоми
- проміжний: все інші
Групи ризику по WPSS
- дуже низький ризик 0 балів
- низький ризик 1 бал
- проміжний ризик 2 бали
- високий ризик 3-4 бали
- дуже високий ризик 5-6 балів

Лікуваннямієлодиспластичного синдрому
Лікування пацієнта з мієлодиспластичним синдромом завжди індивідуально і залежить від:
- симптомів
- стадії захворювання
- віку
- супутніх захворювань
- факторів ризику
- наявності сумісного донора
Лікування пацієнтів з низьким і середнім-1 ризиком мієлодиспластичного синдрому (IPPS 0-1)
Мієлодиспластичний синдром низького і середнього-1 ризику при відсутності змін в аналізі крові не потребує лікування, лишеперіодичне дослідження загального аналізу крові, для виявлення змін в максимально ранній фазі.
- підтримуючу терапію для усунення симптомів — переливання крові, тромбоконцентрату , лікування бактеріальних інфекцій, препарати еритропоетину, стимулятори гранулоцитів (філграстим), тромбоцитів (роміплостім).
- імуносупресори препарати — пригнічують імунну систему, використовуються в лікуванні багатьох захворювань. При мієлодиспластичний синдром імуносупресори застосовуються у пацієнтів з цитопенії при безуспішності або відсутності показань до лікування факторами зростання. Препарати:
- антімітоцітарний глобулін
- циклоспорин
- глюкокортикоїди
- і їх комбінації
- леналідомід рекомендований для лікування мієлодиспластичного сіндромома з ізольованою делецией 5q. Побічні ефекти — свербіж шкіри, висип, диспепсія, втома і підвищений ризик тромбозу.
- Алогенна трансплантація кровотворних клітин

Лікування миелодиспластического сіндромома низького і середнього-1 ризику пересадкою стовбурових клітин крові не входить в стандартний режим. Але, може бути використано у пацієнтів до 60-65 років з важкої і прогресуючої цитопенії, зниженою клітинної кісткового мозку (гіпоплазії) або наявністю сполучнотканинних волокон (фіброзу) в кістковому мозку.
При вторинної формі мієлодиспластичного синдрому (після хіміо- або радіотерапії) трансплантація необхідна, оскільки це МДС високого ризику.
Лікування пацієнтів із середнім-2 і високим ризиком мієлодиспластичного синдрому ( IPPS 1,5-2)
в даній групі вище ймовірність переходу мієлодиспластичного синдрому в гострий лейкоз, тому основна мета — віддалити прогресування захворювання і поліпшити тривалість життя.
- індукційна хіміотерапія — інтенсивна хіміотерапія комбінацією цитостатичних препаратів (тих, що застосовують в лікуванні гострих лейкозів). Умови індукційної терапії при МДС: вік до 65 років, понад 10% бластів у кістковому мозку, без супутніх важких захворювань (хвороби серця, легенів, печінки). Супроводжується багатьма побічними ефектами:
- випадання волосся
- дефекти на слизових оболонках
- нудота
- втрата апетиту
- блювота
- пронос
- висока ймовірністьприєднання бактеріальних ускладнень
- Імовірність ремісії при індукційної хіміотерапії 30-50% в залежності від підвиду мієлодиспластичного синдрому. Після індукційної хіміотерапії, як правило, слід хіміотерапія завершальна, хоча ризик рецидиву в перший рік закінчення лікування досить високий. У даній групі завжди оцінюється можливість подальшої алогенної трансплантації кісткового мозку.
- низькодозового хіміотерапія — низькі дози цітозінарабінозіда (цитозар, Алексан)
- гіпометіляціонние препарати та інгібітори гистон деацетази
- азацітідін — Відаза
- децітабін — Дакоген
мієлодиспластичний синдром у дітей
у дітей мієлодиспластичний синдром зустрічається рідко,0,5-1,5 нові випадки на 1 млн. Дітей, що становить до 5% гематологічних новоутворень. Як і у дорослих, може бути первинним і вторинним. Вторинна форма розвивається після лікування цитостатиками, опромінення, при хвороби Дауна, вроджених аномаліях кісткового мозку (анемія Фанконі). З огляду на несприятливий прогноз у дітей з мієлодиспластичним синдромом на першому місці в лікуванні завжди знаходиться трансплантація кісткового мозку.
Хвороба фон Віллебранда причини, класифікація, симптоми, діагностика, лікування
Хвороба фон Віллебранда — вроджене захворювання c підвищеною схильністю до кровотеч різного ступеня тяжкості, викликане зниженням концентрації, аномалією в будові або функції фактора фон Віллебранда (vWF) .
Історія
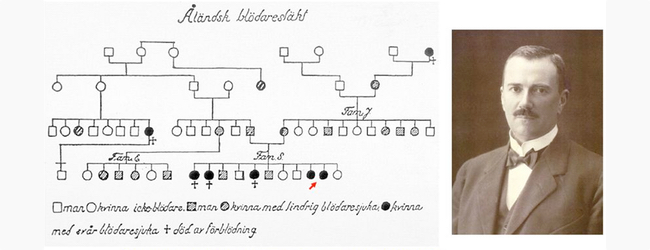
Фінський лікар Ерік Адольф фон Віллебранда в 1924 році описав підвищену схильність до крововиливів в шкіру і слизові оболонки у 5-річної дівчинки і її старших сестер на Аландських островах. Перша пацієнтка, вперше оглянута у віці 5 років, померла після 4-й менструації в 13 років.
Віллебранда достовірно відрізнив нове захворювання від гемофілії і назвав його «вродженої псевдогемофіліей» .
Патофізіологія хвороби фон Віллебранда
— великий білок плазми (15 кДа), бере участь в зупинці кровотечі — активує і стабілізує . Одночасно , підвищується при будь-якому запальному процесі.
Фактор фон Віллебранда синтезують:
- клітини внутрішньої поверхні судин — ендотелій, в них же фактор депонується у вигляді тілець Вейбеля-Паладі
- мегакаріоцити — гігантські материнські клітини тромбоцитів, знаходяться в червоному кістковому мозку
- альфа-гранули тромбоцитів
При попаданні в кров фактор фон Віллебранда розщеплюється металопротеазой ADAMTS-13 і з гігантською молекули виникають менші (моно- і димери).
Фактор фон Віллебранда один з небагатьох білків крові, який має на своїй поверхні аглютиноген групи крові AB0. Саме група крові відповідальна за рівень фактора фон Віллебранда — мінімум у осіб з першою групою крові, максимум — у четверний групи.
Функція фактора фон Віллебранда
- стабілізує фактор VIII (антигемофільних глобулін) , охороняючи його від деактивації протеазами (протеїн С або фактор Xa)
- локалізує фактор VIII в місці кровотечі і формування тромбу
- забезпечує первинний гемостаз — склеювання тромбоцитів між собою (агрегація) і до стінки пошкодженогосудини (адгезія)
При дослідженні фактора фон Віллебранда необхіднооцінити його кількість (антиген) і функціональну активність (ристоцетин кофактор).
Епідеміологія хвороби фон Віллебранда
Хвороба фон Віллбранда — найбільш часте захворювання з підвищеною кровоточивістю. Частота в популяції близько 1%, але симптоми спостерігають у одного з 40 тисяч населення, а небезпечне для життя кровотечу буває у 0,5-3 пацієнтів з 1 млн.
Виділяють дві форми хвороби — вроджену і придбану.
Вроджена хвороба фон Віллебранда
Вроджена хвороба фон Віллебранда викликана мутацією в одному або декількох генах відповідальних за синтез і функцію фактора фон Віллебранда , його взаємодія з тромбоцитами, ендотелієм, фактором VIII. Хворіють як чоловіки, так і жінки.
Відомо більше 300 мутацій на 12-й хромосомі (де закодована молекула vWF), характер яких і визначає тип хвороби фон Віллебранда.
При аутосомно-домінантному спадкуванні симптоми виражені в меншій мірі, але у всіх поколінь. Аутосомно-рецісівний тип проявляється тільки в гомозиготною формі (обидва гена, від обох батьків несуть мутацію) сильними кровотечами.
Класифікація вродженої хвороби фон Віллебранда
- тип 1 — кількість vWF не суттєво знижено (до 15%), функція збережена, аутосомно-домінантний тип спадкування, найбільш частий 80% випадків
- тип 2 — якісний дефект високомолекулярного фактора фон Віллебранда, успадкування переважно аутосомно -домінантное, 15-30% випадків
- тип 3 — повна відсутність фактора фон Віллебранда (0-5%), успадкування аутосомно-рецісівное, вкрай важка форма, фактор VIIII 1-10%, часто діагностують у дітей через симптомів
Придбана хвороба фон Віллебранда
Придбана хвороба фон Віллебранда становить 1% від загального числа випадків.Є наслідком інших захворювань.
- , ( і неходжкінська лімфома), , , — vWF абсорбується на поверхні пухлинних клітин
- синдром Гейден — руйнування vWF при аортальному стенозі, що призводить до , число великих мономерів знижується, що призводить до зниження загальної ефективності фактора
- — споживання vWF
- легенева гіпертензія або пороки серця- VWF швидко розпадається
- (наприклад, ) і — утворюються антитіла до vWF
- — знижена функція веде до недостатнього синтезу фактора в ендотелії
- — рідкісна форма пухлин нирок у дітей, яка поглинає фактор з крові
- лікарські препарати — вальпроєва кислота, ципрофлоксацин, Гекодез
Симптоми хвороби фон Віллебранда
Хвороба фонВіллебранда проявляється різними варіантами кровотеч:
- з носа
- після видалення зубів
- при пораненні (при операції або травми)
- з ясен
- після операції
- після пологів
- шлунково-кишкової кровотечі
- петехии
- гемартрози — крововиливи в порожнину суглобів
- кров у сечі —
- крововилив в головний мозок
Легкі форми захворювання, як правило, сприймаються пацієнтами як звичайнасімейна підвищена схильність до крововиливами.
Хвороба фон Віллебранда — це не «a21> коагулопатия , оскільки фактор фон Віллербанда безпосередньо не бере участь в процесі згортання крові.

Діагностика фон Віллебранда
Діагноз хвороби фон Віллебранда вважається підтвердженим на підставі наступних критеріїв:
- позитивний сімейний анамнез (серед родичів були випадки хвороби)
- схильність до більш частих кровотеч протягом усього життя
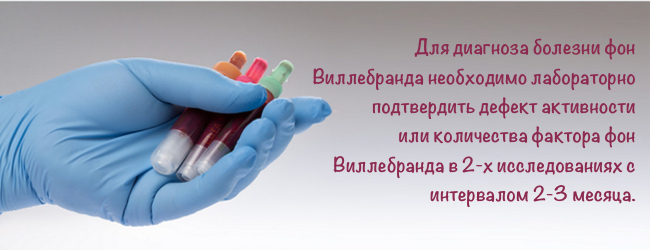
- лабораторно доведений дефект активності або кількості фактора фон Віллебранда в 2-х дослідженнях з інтервалом 2-3 місяці
Аналізи прихвороби фон Віллебранда
- — зниження числа тромбоцитів і в разі хронічного чи гострого кровотечі
- час кровотечі по Лі-Уайту, Дюке, Simplate R (підвищена при 2А, в і М типах)
- згортання крові — видовжене при вторинному дефіциті фактора VIII, в нормі
- фактор VIII в нормі або незначно знижений — для диференціальної діагностики з гемофілією А
- антиген фактора фонВіллебланда (vWF: Ag) — загальна кількість фактора в крові
- функція фактора фон Віллебранда (FVIIIR: Rco) — здатність vWF приєднуватися до активованим глікопротеінові комплексам тромбоцитів, перевіряється адгезія (GP Ib) тромбоцитів після додавання рістоцетін
- зв'язує здатність vWF з фактором VIII
- зв'язує здатність vWF з колагеном (vWF: CBA)
- молекулярно-генетичне дослідження — виявленням мутації (типи 2N і 3) — в
- дослідження мультімеров фактора фонВіллебранда — оцінка різних категорій мультімеров фактора щоб відрізнити підтипи типу 2
Додаткові аналізи при хворобі фон Віллебранда
- — (, , , , ), (, , )
- в
- — , , , , , , , , , активований час рекальцифікації плазми, час кровотечі, час згортання, розчинні фібрин-мономерні комплекси
у легких випадках хвороби фон Віллебранда змін в аналізах може і не бути.
Лікування хвороби фон Віллебранда
легкі випадки хвороби фон Віллебранда не потребують лікування.
Підвищити рівень фактора фон Віллебранда в крові можна двома шляхами:
- активувати виділення ендогенного (внутрішнього, запакованого в ендотелії і тромбоцитах) vWF
- подати фактор ззовні
Десмопрессин або DDAVP (1-деаміно-8-D-аргінін вазопресин) — синтетичний аналог , вивільняє vWF з ендотелію судин. Лікувальна доза 0,3 мкг / кг внутрішньовенно з максимальним підвищенням vWF і фактора VIII через 30-60 хвилин після закінчення введення. Ту ж дозу можна ввести і підшкірно з максимумом через 60-120 хвилин. Інтраназальне введення (через ніс) по 150 мкг в кожну ніздрю (при вазі тіла до 50 кг — дозу зменшити у 2 рази), максимум vWF через 60-90 хвилин. Введення вазопресину підвищує vWF в 3-5 разів. Повторні аплікації знижують ефект на 30%, тому інтервал повинен бути не менше 12-24 годин до 5 днів. Ефективність лікування десмопресином також залежить від типу хвороби фон Віллебранда. Тип 1 — ефект максимальний, тип 2А і 2М менший, а при 2В ще більш знижує число тромбоцитів, тому протипоказаний. При 3 типі — таке лікування безуспішно.
Очищений плазмовий концентрат фактора VIII з vWF та високоочищений концентрат vWF (з або без фактора VIII) використовують для заповнення дефіциту vWF.
Стандарт замісного лікування хвороби фон Віллебранда — концентрат фактора VIII з vWF ( 1: 2,2) препарат Haemate P (гематом П, Behring). Інші аналогічні препарати даної групи для лікування хвороби фон Віллебранда,
- Fahndi (Grifols)
- Alfanate (Grifols)
- Immunate (Baxter)
- VHP-vWF (LFB)
антифібринолітиками прапарати — трексановая кислота, 25 мг / кг кожні 8 год при дрібних крововиливах в слизові оболонки, кровотечі з носа, менструальних кровотечах.
Рекомендовано переливання тромбоконцентрату при зниженні числа тромбоцитів і гормональні протизаплідні препарати при великих втратах крові при менструації. Етамзилат застосовують для зупинки незначних кровотеч.
Діагностика алергії — алергічні проби
Алергічні проби
для виявлення алергічного захворювання і визначення алергену використовують методи in vitro — в пробірці , т . Е. в лабораторії і in vivo — на живому організмі самого пацієнт а .
Вся складність полягає в тому, щоб зробити правильний вибір методу дослідження в діагностиці алергії і надати ефективну та своєчасну допомогу хворому. У багатьох випадках лабораторний аналіз показує наявність схильності до алергії у пацієнта (наприклад, наявність імуноглобуліну Е (IgE) проти кліщів), але захворювання (риніту або астми) не спостерігається.
Для підтвердження правильності вибору тієї чи іншої алергічної проби необхідно вивчити анамнез пацієнта (історію життя, захворювання та ін.) або в деяких випадках провести провокаційний тест.
Підготовка до алергічної пробі
Згода пацієнта
Пацієнт повинен бути згоден на проведення проби, в іншому випадку процедуру слід скасувати. Після того як пацієнт поінформований про ступінь ризику заходи, він повинен письмово підтвердити свою згоду.
Вибір ділянки тіла для тестування
Тестування на алергію НЕ має проводитися на області, схильною до запалення, або там, де раніше відзначався запальний процес. Пацієнтам з кропив'янкою, викликаної фізичним навантаженням, протипоказано проведення проб на I тип алергічних реакцій — негайного типу.
Протипоказання до алергічних проб :
- вагітність
- дитячий вік
- наявність інших важких захворювань
- виявлена активна інфекція в тестованих органах
- використання імунодепресантів
- використання препаратів, які можуть спотворити результати проби (інгібітори АПФ — препарати для зниження тиску — каптоприл, еналаприл, лізиноприл та ін.)
- поточні алергічні проблеми
- ймовірність важкої, небезпечної для життя реакції на тестові речовини.
на які речовини проводяться алергічні проби?
Тест-з'єднання на алергію являють собою набір стандартних алергенів.
Екстракт алергенів перевіряється за допомогою специфічних антитіл з метою кількісного визначення алергену методом ELISA. Кількості різних алергенних білків виявляються методом електрофорезу. Рекомбінантні антигени також можуть використовуватися, якщо їх імуноглобулін Е (IgE) -зв'язуючим ділянку ідентичний природному.
Все новосинтезовані тест-з'єднання на алергію повинні відповідати наступним характеристикам:
- відсутність дратівної або токсичної дії
- ризик інфікування виключений
- проведена контрольна апробація на добровольцях.
Тест-методи на алергію
тест-методи на алергію повинні бути стандартизовані. Проба уколом (прик-тест) повинна проводитися з дотриманням наступних умов:
- проби проводяться на внутрішній поверхні передпліччя
- проби вводяться на відстані 2 см
- дотримується постійний проміжок часу від уколу до аналізу отриманого результату
- застосовуються стандартні методи оцінки, такі як наявність еритеми і пухиря за шкалою від + до + + +.
У кожній окремій методиці тестування шляхом проби уколом можуть використовуватися додаткові стандартні шкали.
В інших органах більш прийнятна функціональна оцінка (наприклад, дихальна функція).
Ускладнення алергічних проб
Шкірні та системні провокаційні тести на алергію спрямовані на те, щоб з'ясувати, чи буде імуноглобулін Е — опосередкована реакція гіперчутливості негайного типу викликати більш важкі реакції, такі як кропив'янка, астма та анафілаксія.
Поява подібних реакцій говорить про високу чутливість пацієнта до аналізованого алергену (проба позитивна).
Через ризик розвитку анафілаксії алергічні проби проводяться тільки досвідченими професіоналами. Весь персонал також повинен бути підготовлений і в разі критичного стану діяти згідно з інструкціями.
Підготовка до проведення алергічної проби включає в себе і розробку плану надання швидкої медичної допомоги, якщо це трапиться в офісі або віддаленій від центру клініці, наявність необхідного набору медикаментів швидкої допомоги і навчання персоналу.
В тих випадках, коли ймовірність розвитку анафілактичної реакції велика, наприклад, при тестуванні на алергію до бджолиним або осиним токсинів, необхідно вставити катетер у вену перед початком проби і збільшити період спостереження після проведення проби. Тестування таких пацієнтів слід проводити в лікарняних умовах.
Можливі помилки в алергічних пробах
Незважаючи на те, що основна помилка вже була описана вище в статті, необхідно повторитися, що повинна бути встановлена клінічна значимість всіх позитивних результатів тестування . в іншому випадку пацієнт буде дотримуватися дієти або піддаватися лікуванню, в яких немає необхідності.
Вплив психологічних факторів також може привести до хибнопозитивного відповіді на алергічну пробу.
Так, наприклад, при тестуванні на харчову алергію виявлена певна проблема, в цьому випадку вважається ефективним застосування плацебо-контролю і подвійного сліпого методу.
Лабораторна діагностика алергії
Звичайні алергічні тести в умовах лабораторії полягають в дослідженні крові або інших рідин організму (наприклад, назального секрету) з метою оцінки імунологічної реактивності. Лабораторні тести на алергію можуть використовуватися для скринінгу алергії і виявлення потенційних алергенів.
У деяких випадках аналізи застосовуються на додаток до шкірних тестів уколом або ж замінюють їх, особливо якщо є такі протипоказання до проведення останніх:
- пацієнт — маленька дитина
- у пацієнта велике шкірне захворювання і не може бути вибраний вільну ділянку шкіри для тестування
- у пацієнта існує ризик розвитку анафілаксії при проведенні шкірних проб
- рідко зустрічається алерген, стандартний розчинякого в даний момент недоступний
- пацієнтові не можна припиняти поточну антигистаминную терапію.
Проведення аналізів на алергію
Імуноглобулін Е ( IgE )
Для вимірювання кількості , що виділяється у відповідь на вплив специфічного антигену, існує кілька методів:
- простий метод напівкількісного аналізу, такий як тест-смужки або метод іммунодота, при якому алерген точково наносять на нітроцелюлозні смужки
- складніші процедури,такі як радіоаллергосорбентний тест (RAST) або твердофазного імуноферментного тест (ІФА, ELISA).
В основі всіх цих тестів лежить один і той же принцип. Тестований антиген фіксується на носії (тест-смужка або диск для алергічних проб). Тестовані сполуки инкубируются разом з сироваткою пацієнта, яка може містити антитіла до алергену, що призведе до утворення комплексу антиген-антитіло. Ці комплекси метятся анти-IgE антитілами, після чого їх можна ідентифікувати за допомогою хромогенного речовини або радіоактивних міток шляхом вимірювання на сцинтиляційному лічильнику.
Загальний імуноглобулін Е
Вимірювання імуноглобуліну Е (IgE) допомагає лише зорієнтуватися в ситуації, коли йде пошук передбачуваного алергену. У нормі вміст IgE в сироватці крові 17-240 нг / мл (2,4 нг / мл = 1 МО / мл). Якщо кількість імуноглобуліну Е IgE перевищує 170 нг / мл (70 МО / мл), можна підозрювати наявність алергії, однак у багатьох здорових людей цей показник набагато вищий, і вони не мають на алергії, ні атопії.
У деяких пацієнтів, які зазвичай страждають алергічним або атопічний захворюванням, цей рівень перевищує 10 000 МО / мл. Підвищений рівень IgE виявляють також при:
- ,
- ,
- вродженому імунодефіциті,
- імуносупресії — пригніченні імунітету,
- реакції відторгнення трансплантата,
- при важких опіках.
Зазвичай рівень IgE вимірюють за допомогою методу ELISА (ІФА).
Мультитест
При скринінгу алергенів найбільш зручні системи, в яких різні алергени пов'язані з одним носієм. З цією метою використовуються як диски, так і розчини, що містять алергени. Даний тест дуже економічний і зручний.
Якщо в лабораторних тестах на алергію не виявлено вираженої сенсибілізації, необхідно використовувати інші, більш прямі методи ( уколом, визначення ).
Тест-смужки містять менше антигенів і менш точні.
Специфічний імуноглобулін Е
Кількісне визначення специфічного імуноглобуліну Е (IgE, один з видів ), спрямованого проти певного антигену, проводять за допомогою RAST або інших подібних тестів. При цьому можна перевірити до 500 алергенів.
Для кожної проби необхідно 50 мкл сироватки або секрету. Для контролю точності аналізу і сили відповіді використовують стандартну сироватку пацієнтів з алергією. Змісту IgE відносять до одного з класів по 4-6-бальній, але між концентрацією IgE і ступенем прояву клінічних симптомів немає прямої залежності.
Еозинофільний катіонний білок (ЕСР)
Катіонний білок еозинофілів (ЕСР) вивільняється з активованих еозинофілів в результаті їх руйнування в ході алергічної реакції, він володіє цитотоксическим і нейротоксическим дією. У нормі, вільний ЕСР можна виявити в крові.
Кількість ЕСР відображає ступінь активації еозинофілів, однак його рівень визначають в крові, що згорнулася, в якій еозинофіли піддаються руйнуванню. У нормі кількість ЕСР має бути менше 105 нг / мл.
При деяких захворюваннях в крові та інших рідинах організму визначається підвищений вміст ЕСР:
- реакції гіперчутливості негайного типу
- паразитарні інфекції
- неаллергический риніт з еозинофільним синдромом (NARES)
- назальні поліпи.
Рівень ЕСР — дуже варіабельний показник і не завжди підвищений при алергічних ринітах. При антиалергійною терапії, наприклад при прийомі кортикостероїдів, зазвичай спостерігається тенденція до зниження рівня ЕСР, що дозволяє простежувати перебіг хвороби.
триптази вивільняється із стовбурових клітин в процесі реакцій типу I — негайні реакції, наприклад , при анафілактичний шок. Вона більш стабільна, ніж гістамін, і легше ідентифікується.
Фізіологічні функції триптази не зовсім вивчені, але очікується, що вона, як і калікреїн, бере участь в синтезі брадикініну.
Тест дегрануляции баз офілов ( BDT )
Тест дегрануляции базофілів ( BDT ) також відомий як тест на вивільнення гістаміну.
з крові пацієнта виділяють базофіли і змішують їх в тест-пробірці з передбачуваним алергеном протягом 30 хвилин. Далі вимірюють вміст гістаміну. Як позитивного і негативного контролів аналогічну процедуру проводять з кров'ю пацієнтів-алергіків і здорових людей.
Базофіли пацієнтів з алергічним захворюванням досить чутливі і часто процес дегрануляції носить неспецифічний характер.

Тест стимуляції клітин алергеном ( CAST ) аналогічний тесту BDT, але в цьому випадку вимірюють кількість лейкотрієнів (D4, C4 і Е4), вивільнених з лейкоцитів . Так, як ці продукти синтезуються заново після впливу специфічного алергену, то неспецифічна стимуляція навряд пі надасть дію на результати тесту.
CAST є більш точним тестом в порівнянні з вимірюванням специфічного імуноглобуліну Е (IgE) і застосовується тільки для ідентифікації реакцій типу I — негайних реакцій.
іммунодіффузія (метод Ухтерлоні) використовується для виявлення таких антитіл, як антитіла проти сироватки гопубя (легкі любителя птахів) і антитіла проти Aspergillus ( легеневий аспергільоз).
Тестована сироватка наноситься в лунку в центр чашки з агаром, а антигени в різних концентраціях поміщають в лунки навколо. Антигени і антитіла починають дифундувати через гель у напрямку один до одного.
У разі присутності в тестованої сироватці специфічних антитіл, починають формуватися комплекси антиген-антитіло, створюючи лінію преципітації.

перехресний іммуноелектрофорез ( CIE ) і перехресний радіо іммуноелектрофорез ( CRIE )
CIE (перехресний іммуноелектрофорез) і CRIE (перехресний радіоіммуноелектрофорез) застосовують для ідентифікації та стандартизації екстрактівалергенів, які використовуються для шкірної проби уколом, провокаційних тестів і гипосенсибилизации. CIE використовується для визначення різновидів антигену, a CRIE — для визначення антигену.
Обидва тести можуть використовуватися для моніторингу гипосенсибилизации.
Атопічний дерматит
Термін атопічний дерматит походить від грецького слова atopos — невідповідний. Це захворювання належить до атопічний алергій — Атопія , до яких також відносяться алергічна астма і алергічний рінокон'юнктівіт.
Пацієнти з атопией мають спадкову схильність до розвитку неадекватної алергічної реакції на первинні природні алергени. У родині одна людина може страждати на атопічний дерматит, інший — алергічної астмою.
Дуже часто у пацієнтів з атопічним дерматитом спостерігається підвищений рівень специфічного і загального імуноглобуліну Е (IgE). Часто перебіг атопічного дерматиту має циклічний характер і залежить від умов навколишнього середовища та соціальних факторів.
Симптоми атопічного дерматиту
прояви атопічного дерматиту дуже різні в залежності від віку пацієнта.
Симптоми атопічного дерматиту у дітей до 10 років
Атопічний дерматит у немовлят — дитяча екзема, зазвичай розвивається в тримісячному віці і відноситься до найбільш поширених атопічним захворювань.
Хворіють 5-10% дітей, що є першою ознакою майбутніх атопічних захворювань у дитини. У дитячому віці атопічний дерматит протікає в гострій формі і з великою кількістю ексудату, зазвичай поширюючись на обличчя, голову, тулуб і згини кінцівок. Він супроводжується свербінням і частим виникненням вторинної інфекції — золотистого стафілококу ( Staphylococcus aureus ).
на жаль, не існує будь-якого високоефективного лікування, мета призначень — максимально полегшити стан дитини і запобігти ускладненням.
Зазвичай позитивний момент в перебіг цієї хвороби настає у віці 2-4 роки, коли більшість дітей одужують.

Атопічний дерматит у підлітків
Через роки (з початком навчання в школі, наступ пубертатного періоду) атопічний дерматит може виникнути знову. Тоді захворювання приймає хронічну форму, ліхенізація — потовщення шкіри проявляється сильніше, зачіпаючи в основному потилицю і згини кінцівок.
Шкіра виглядає пересушеній, в дрібних лусочках і дуже чутлива до найменшого подразнення (наприклад, до вовняному одязі). Така форма дерматиту, на яку страждає приблизно 1-3% підлітків, найчастіше зникає в юнацькому віці.
Лише у незначної кількості пацієнтів дерматит зберігається в хронічній формі, причому проблеми можуть виникати протягом усього життя, висловлюючись в подразненнях і кумулятивному токсичному дерматиті.
Таким людям необхідно дуже серйозно поставитися до вибору професії і забезпечувати додатковий догляд за шкірою, якщо вони піддаються впливам вологи і потенційних дратівливих чинників (наприклад, при професійній зв'язку з косметологією або процесами приготування їжі).
Атопічний дерматит у дорослих
Третя фаза атопічного дерматиту може наступити у віці близько 30 років, але іноді і в 60 років.
У дорослих дерматит супроводжується сильним свербінням, поширюється широко, визначаються локальні осередки сильно ороговілих папул і вузликів (пруріго) на тілі і згинах кінцівок, проте іноді у дорослих пошкоджується тільки шкіра на повіках, руках або потилиці.
Відзначається також підвищена чутливість шкіри, непереносимість вовняних тканин, а також можливість інфікування стафілококом .
Третя фаза атопічного дерматиту найважче піддається лікуванню.

Патогенез атопічного дерматиту
Патогенез атопічного дерматиту абсолютно незрозумілий і дані досить суперечливі.
Хвороба має багато спільного з алергічною астмою і рінокон'юнктівітом, тому існує припущення, що процес є «алергічну »активацію Т-лімфоцитів. Однак більшість дослідників вважають, що алергічні реакції не є визначальним фактором в патогенезі атопічного дерматиту.
Відповідно до гіпотези про «аутоімунної» формі дерматиту , він являє собою інтерлейкін 4 (IL-4) опосередковану алергію з підвищеним рівнем імуноглобуліну Е (IgE), який ініціює процес презентації аутоантигенов Т- лімфоцитів за допомогою клітин Лангерганса в шкірі.
Найважливіші діагностичні критерії атопічного дерматиту
- хронічний дерматит, виявляється на певних зонах, у відповідному віці
- наявність атопії в сім'ї
- сухість і подразливість шкірних покривів
- алергічний рінокон'юнктівіт і / або астма
- гіперлінеарность долонь і стоп
- підвищений рівень специфічного або загального імуноглобуліну Е (IgE).

Лікування атопічного дерматиту
Незважаючи на постійні пошуки альтернативної терапії для купірування гострих симптомів атопічного дерматиту, основними препаратами залишаються кортикостероїди .
Активно розглядається можливість терапії атопічного дерматиту за допомогою макролідів в якості імуномодуляторів. Ці імуносупресори препарати досить ефективні, але їх відстрочені побічні ефекти невідомі.
Найважливіше забезпечувати ретельний догляд за шкірою, приділяти особливу увагу її зволоженню і пом'якшенню, а також проводити своєчасну терапію можливих стафілококових інфекцій.
Фактори, що сприяють розвитку атопічного дерматиту
Хоча причина виникнення атопічного дерматиту до сих пір не ясна, не викликає сумнівів, що в його основі лежить взаємодія генетичних і зовнішніх чинників. У новонароджених грає роль харчова алергія (яйця, молоко), у дітей старшого віку більш важливою вважають алергію на вдихувані речовини.
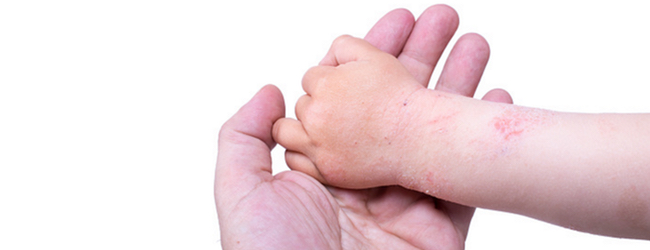
Зазвичай шкіра таких дітей легко пошкоджується після контакту з дратівливими речовинами, а також при сповивання в результаті здавлювання. Пізніше велике значення мають носіння вовняного одягу і неадекватний догляд за шкірою, наприклад, занадто часте миття і використання великої кількості мила.
Патогенез атопічного дерматиту у дітей
Єдиної моделі патогенезу атопічного дерматиту не існує, є лише безліч індивідуальних спостережень.Можливо, має важливе значення імунологічний дисбаланс з домінуванням активності Т2-хелперів.
Крім того, в патогенезі атопічного дерматиту важливу роль відіграють зв'язують імуноглобулін Е (ІgЕ) — антигенпрезентуючими клітини шкіри, які здатні ініціювати «аутоіммунну» реакцію проти шкіри .
При атопічний дерматит часто можна спостерігати алергію типу I (негайного типу), а на більш пізніх стадіях в результаті неправильного догляду за шкірою і лікування — реакції типу IV.
Симптоми атопічного дерматиту у дітей
Клінічні прояви атопічного дерматиту у дітей вкрай різноманітні і змінюються з віком пацієнта. Дерматит завжди супроводжується:
- еритемою — почервонінням шкіри,
- дрібними пухирями,
- освітою лусочок
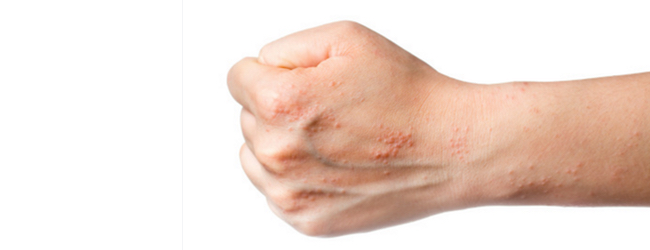
Якщо атопічний дерматит прогресує, неминуче освіту корости.
У маленьких дітей першою ознакою дерматиту часто є поява еритематозних набряку обличчя, а іноді шиї і кінцівок. Через дуже сильного свербежу дитина ставати пригніченим і роздратованим, так як його шкіра постійно свербить.
У дітей шкільного віку дерматит зазвичай захоплює кінцівки, включаючи ліктьову і підколінну складки і шию, проявляючись у вигляді ліхенізірованной — потовщених утворень і тріщин на шкірі.
При атопічний дерматит у дітей дуже поширене поява вторинної інфекції, в 90% випадків це золотистий стафілокок ( Staphylococcus aureus ). часто виникають вірусні інфекції, які протікають швидкоплинно і важко, наприклад, при вірусі простого герпесу (герпетична екзема) або поксвирусов (контагіозний молюск, eczema vaccinatum при імунізації проти віспи).
Диференціальна діагностика виявляє коросту і синдром гіпер — I gE, який розглядається як рідкісний генетично детермінований максимальний варіант атопічного дерматиту.
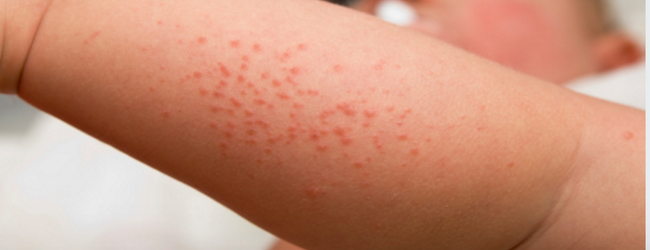
Лікування атопічного дерматиту у дітей
На 100% ефективного лікування атопічного дерматиту у дітей не існує .
Необхідний самий ретельний догляд за шкірою.
Золотим правилом в лікуванні атопічного дерматиту у дітей є наступне:
- якщо шкіра суха, обробку необхідно проводити з допомогою емульсійних кремів і мазей,
- якщо жирна, потрібно використовувати зволожуючі розчини і не забивають пори креми або лосьйони.
Допомагає при атопічний дерматит у дітей застосування ванн з зволожуючими маслами або обробка ними після процедури. Необхідно уникати дратівливих впливів.
Ліки в лікуванні атопічного дерматиту у дітей
Оптимальним при атопічний дерматит у дітей є короткочасний курс (3-5 днів) місцевих кортикостероїдів, що дозволяє взяти під контроль поточний процес і уникнути ускладнень. Однак умовити батьків використовувати ці препарати часто буває складно через острах «кортизону».
Так як часто виникають вторинні інфекції, показано застосування коротких курсів антибіотиків або місцевих антисептиків (що не викликає роздратування).
У разі герпетичної екземи у дитини необхідно використання системних вірусостатіческім препаратів.
Виключення подразників
виявити і потім уникати дратівливих агентів при атопічний дерматит у дитини — це складне завдання. У дітей старшого віку хороший ефект дає гипосенсибилизация.
Кліматична терапія протягом не менше чотирьох тижнів також може бути корисна. Однак незаперечних доказів тривалого ефекту цих методів при атопічний дерматит у дітей немає. При цьому негативним побічним ефектом може бути стрес через зміни звичних умов перебування в школі і сім'ї.